分析化学实验思考题总结
实验一 煤气灯的使用和玻璃工操作
1. 正常的煤气灯火焰,各焰层的大概温度为多少?被加热的物体应放在哪一层?(题目加黑)
答:焰心(内层)温度约573K左右;还原焰(中层)温度较焰心高;氧化焰(外层)温度最高,约1073~1173K。实验时,被加热物体一般都用氧化焰来加热,根据需要可调节火焰的大小。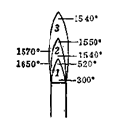
(留一个空)
2.使用煤气灯时,什么情况下会出现临空火焰和侵入火焰?出现这种情况如何处理?
答:如果煤气和空气的进入量都调节得很大,则点燃煤气后火焰在灯管的上空燃烧,移去点燃所用的火柴时,火焰也自行熄灭,这样的火焰称为“临空火焰”。 如果煤气的进入量很小,而空气的进入量很大时,煤气将在灯管内燃烧,管口会出现一缕细细的呈青色或绿色的火焰,同时有特色的“嘘嘘”声响发出,这样的火焰称为“侵入火焰”。 遇到这些不正常的火焰,应立即关闭煤气开关,重新调节和点燃煤气。
3.选择瓶塞有什么要求?试比较玻璃磨口塞、橡皮塞和软木塞各有哪些优缺点。
答:塞子的大小应与仪器的口径相适合,塞子塞进瓶口或仪器口的部分不能少于塞子本身高度的1/2,也不能多于2/3。瓶塞的种类应根据所装化学品的化学性质来选择。软木塞易被酸、碱所损坏,但与有机物作用较小。橡皮塞可以把瓶子塞得很严密,并可以耐强碱性物质的侵蚀,但容易被强酸和某些有机物质(如汽油、苯、氯仿、丙酮、二硫化碳等)所侵蚀。玻璃磨口塞子把瓶子也塞得很严,它适用于除碱和氢氟酸以外的一切盛放液体或固体物质的瓶子。
4.将玻璃管插入塞孔时,应如何操作?
答:将选定的玻璃导管插入并穿过已钻孔的塞子,一定要使所插入导管与塞孔严密套接。
先用右手拿住导管靠近管口的部位,并用少许甘油或水将管口润湿,然后左手拿住塞子,将导管口略插入塞子,再用柔力慢慢地将导管转动着逐渐旋转进入塞子,并穿过塞孔至所需的长度为止。也可以用布包住导管,将导管旋入塞孔。如果用力过猛或手持玻璃导管离塞子太远,都有可能将玻璃导管折断,刺伤手掌。
5.如何确定火焰的不完全燃烧(可借助于仪器和物品)?
答:点燃煤气灯时煤气燃烧不完全,便会析出碳质,生成光亮的黄色火焰,且火焰温度不高。
6.加热玻璃管时,应如何使玻璃管受热均匀?
答:先将玻璃管用小火预热一下,然后双手持玻璃管,把要弯曲的部位斜插入喷灯(或煤气灯)火焰中,以增大玻璃管的受热面积(也可在灯管上罩以鱼尾灯头扩展火焰,来增大玻璃管的受热面积),若灯焰较宽,也可将玻璃管平放于火焰中,同时缓慢而均匀地不断转动玻璃管,使之受热均匀。
7.实验室使用的塞子一般有玻璃塞、橡皮塞、软木塞,分析各种塞子有什么优缺点。
答:软木塞易被酸、碱所损坏,但与有机物作用较小。橡皮塞可以把瓶子塞得很严密,并可以耐强碱性物质的侵蚀,但容易被强酸和某些有机物质(如汽油、苯、氯仿、丙酮、二硫化碳等)所侵蚀。玻璃磨口塞子把瓶子也塞得很严,它适用于除碱和氢氟酸以外的一切盛放液体或固体物质的瓶子。
实验二 滴定分析基本操作(加上题目,有些题目回答太简单)
1、1:1的盐酸是指1体积的浓盐酸(质量分数:36%~38%)与1体积的水混合配比而成,摩尔浓度约为6.25mol/l。
2、强碱性,强吸湿性,应使用干燥的小烧杯称量。
3、刚洗过的滴定管、移液管的内壁挂有小水珠,直接滴定或移液会增加原溶液的水,从而造成实际滴定液浓度的变化,而产生较大误差。所以要润洗,使挂在内壁上的液体尽量与所配溶液浓度一致,润洗一般至少三次。
锥形瓶不需润洗,因为需要准确不变的酸(或碱)的量,而不是浓度,如果将盛有待滴定酸(或碱)液的锥形瓶润洗了,会使酸(或碱)的量增加,从而造成大的误差。
4、甲基橙的变色范围是pH=3.1~4.4,酚酞的是pH=8.0~10.0。在碱性条件下,甲基橙显黄色,酚酞显橙色;酸性条件下。甲基橙显橙色,酚酞为无色。根据人眼的感觉习惯,当颜色有无色到有色,由浅色到深色,由冷色到暖色变化时,较易判断,所以,酸滴碱时用甲基橙做指示剂,碱滴酸时用酚酞。反之,则滴定终点不易准确判断,易造成较大误差。
5、在以cm3作为单位的情况下,用滴定管、移液管、容量瓶作量器时,记录的数据应精确到小数点后两位。有效数字3~5位。
6、滴定管在制作过程中,难免会出现粗细不均,用同一段管子可以减小仪器误差。
7、这样做,移液管尖端的保留体积会基本相同,不会导致平行滴定时的过大误差。因为一些管口的尖端加工的不十分圆滑,如果不旋转,停顿,会使每次在尖端的保留体积差异过大,而造成较大误差。
8、因为,可能会有溅沾在锥形瓶壁上的滴定液或待滴定液,用去离子水冲洗瓶壁会使两者反应完全,结果更准确。在接近终点时显色剂变色灵敏,稍有不慎便会过量,所以要缓慢滴加。
实验三食盐提纯
1,理论上在室温下,8.0g食盐形成饱和溶液需要22cm3左右的水,但在加热蒸发过程中水会变少,所以加水30cm3。加水过多会使实验时间过长,加水太少会使食盐无法完全溶解或者过早析出。
2,在沉淀的形成的过程中,如果溶液的浓度过大,形成的沉淀颗粒较小,过滤分离的速度就会太慢。逐滴加入是为了SO42-离子不是大量的过量,避免形成的沉淀太小。继续加热是为了陈化的过程,使小颗粒溶解,大的颗粒长大,利于过滤。
3,不可以,因为MgCO3是微溶物,加入OH-才能使Mg2+沉淀完全。加热至沸是为了使它们完全反应。
4,Ba2+由后面加入的Na2CO3反应生成BaCO3沉淀除去,NaOH与Na2CO3由后面加入的HCl除去,氯离子是食盐的组成成分。
5,因为其中还有KCl,必须通过它与NaCl溶解度的不同除去。
6,防止CO32-的干扰,CO32与Ba2+也能形成沉淀,但是沉淀可以被盐酸分解。
7,为了防止溶液中可能仍然存在的CO32-及OH-的干扰。Ca2+与C2O42-形成的草酸钙沉淀可以被盐酸分解而不能被乙酸分解。
8,根据两者溶解度随温度变化的不同,通过重结晶的方法除去,必要的时候可以多次重结晶。
9,普通漏斗主要应该注意滤纸的叠法、滤纸与漏斗的结合要紧密,漏斗颈紧靠在接受容器的内壁上,转移时用倾析法,先转移溶液后转移沉淀,同时用玻璃棒靠在三层滤纸处引流,每次加入的溶液不要超过滤纸高度的2/3。
减压过滤应注意倒吸。漏斗缺口靠近空气出口,过滤完毕先拔漏斗后关抽气设备。
优缺点比较:普通漏斗速度慢,装置简单,不易收集固体。
减压漏斗过滤速度快,固体和液体均可收集。
10,因为盐酸浓度较大,调节pH容易过量,所以应该小心滴入,必要时可以将溶液稀释。
11,CO2溶解在水中的pH约为5,即此酸度下,CO32-被分解,可以除去残留的CO32-。pH试纸的使用:将小块试纸放在干燥洁净的点滴板上,用玻璃棒沾取待测的溶液,滴在试纸上,观察试纸的颜色变化,在30秒内尽快将试纸的颜色与标准比色板颜色对比。
12,这些可能是残留在蒸发皿壁上的氯化钠及其他杂质,这时可以通过加入去离子水使其溶解到溶液中而减少损失。
实验四氯化物中氯含量的测定
1. 莫尔法测氯时,为什么介质的pH须控制为6.5-10.5?
答:在酸性介质中,铬酸根将转化为重铬酸根,溶液中铬酸根的浓度将减小,指示终点的铬酸银沉淀过迟出现,甚至难以出现;若碱性太强,则有氧化银沉淀析出。
2. 以K2CrO4作指示剂时,指示剂浓度过大或过小对测定有何影响?
答:K2Cr2O7的浓度必须合适,若太大将会引起终点提前,且其本身的黄色会影响终点的观察;若太小又会使终点滞后,都会影响滴定的准确度。根据计算,K2CrO4的浓度约为5×10-3 mol/L为宜。
3. 用莫尔法测定“酸性光亮镀铜液”(主要成分为CuSO4和H2SO4)中的氯含量时,试液应做哪些预处理?
答:加入KCN掩蔽Cu2+,并加入适量的NaOH中和硫酸,控制溶液为中性或弱碱性。
4. 若试样中有较多的杂质离子,加入沉淀剂可以形成沉淀,对于测定结果将会有什么影响?讨论莫尔法测氯的适用性。
答:由于指示终点的判断是根据生成砖红色的铬酸银沉淀,若杂质离子的沉淀有颜色会影响终点的判断,并会消耗沉淀剂,使最终的测定结果偏大。
可行性: 在中性溶液中,以K2CrO4为指示剂,用AgNO3标准溶液滴定Cl-,称为莫尔法,滴定反应 Cl-(aq) + Ag+(aq) = AgCl(s) (白色); Ksp(AgCl)=1.8×10-10
终点反应 CrO42-(aq) + 2Ag+(aq) = Ag2CrO4(s)(砖红色);Ksp(Ag2CrO4)=1.2×10-12
由于Ag2CrO4的溶解度比AgCl大,根据分步沉淀的原理,溶液中将首先析出AgCl。当AgCl定量沉淀后,稍过量的Ag+即与CrO42-生成砖红色的Ag2CrO4,使沉淀染上明显的红色。凡能与Ag+和CrO42-生成沉淀或配合物的离子都干扰测定,如PO43-、AsO43-、S2-、C2O42-、Ba2+、Pb2+、Hg2+等,以及在中性或微碱性溶液中易水解的离子,如Fe3+、Al3+等,以及有色离子如Cu2+、Co2+、Ni2+等量大时也都妨碍测定,应预先分离除去。本法终点明晰,结果准确。
5. 测定接近终点时,加水冲洗锥形瓶对测定结果有影响吗?
答:有影响,加水冲洗会稀释溶液,使终点变色不敏锐,甚至使终点推迟,结果偏大。
6. 为何要控制滴定速度?与酸碱滴定比较,控制速率的意义有何不同?
答:因为滴定速度的快慢影响反应进行的快慢及程度,滴定速度过快会造成局部浓度过高,提前到达反应终点,而实际上其它部分尚未反应,使滴定失败。(沉淀的吸附的影响)
在酸碱滴定中,滴定速度过快使局部快速达到中和,导致指示剂颜色变化,而在沉淀滴定中,滴定速度过快使局部快速饱和而生成沉淀,与酸碱滴定相比,控制滴定速率尤为重要,直接决定滴定的成功与否。
7. 使用AgNO3溶液有哪些注意事项?洗涤装过AgNO3标准溶液的滴定管,用自来水冲洗会有什么发现?应该怎样清洗?
答:AgNO3溶液应该放在棕色试剂瓶中,置暗处保存,滴定时要快速装入棕色滴定管,防止见光分解。
洗涤时,先用去离子水冲洗;如果已经发现有白色不溶物,并且不易洗去,可先用适量的氨水冲洗,然后用去离子水冲洗。
实验五混合碱的测定
1. 欲测定混合碱中总碱度,应选用何种指示剂?为什么?
答:应采用甲基橙等做指示剂。因为甲基橙的变色范围为3.1 ~ 4.4,在酸性范围内变色,保证混合碱中无论强碱还是弱碱都参与反应,准确指示终点。
2. 采用双指示剂法测定混合碱,在同一份溶液中测定,试判定下列五种情况下,混合碱中存在的成分是什么?
(1)V1=0 (2)V2=0(3)V1>V2(4)V1<V2(5)V1=V2
答:① V1=0 时,组成为:HCO3-
② V2=0时,组成为:OH-
③ V1>V2时,组成为:CO32-+ OH-
④ V1<V2时,组成为:HCO3- +CO32-
⑤ V1=V2时,组成为: CO32-
3. 无水碳酸钠保存不当,吸水1%,用此基准物质标定盐酸溶液时,其结果有何影响?用此浓度测定试样,其影响如何?
答:吸水后,使盐酸浓度偏大;测定试样,会使氢氧化钠和碳酸钠的含量都增加,但百分含量不变。
4. 测定混合碱时,到达第一化学计量点前,由于滴定速度太快,摇动锥形瓶不均匀,致使滴入的HCl局部过浓,使NaHCO3继而分解为CO2而损失,此时采用酚酞为指示剂,记录V1,如此操作对测定结果有何影响?
答:若混合碱为NaOH和NaCO3,则会使V1增加,V2减小,即NaOH百分含量增加,NaCO3百分含量减小。
若混合碱为NaCO3和NaHCO3,则会使V1增加,V2减小,即NaCO3百分含量增加,NaHCO3百分含量减小。
5. 评价一下双指示剂法的准确性和误差来源,整个过程只用一种HCl溶液而不标定其浓度可以吗?
答:准确性:第一化学计量点pH=8.3,用酚酞为指示剂,从红色滴到无色(微红)。因为酚酞变色范围 pH = 8.0~10.0,pH=8.3时,无色略带一点微红,此时NaOH——完全滴定;Na2CO3——滴到NaHCO3 ;NaHCO3——不反应。第二化学计量点pH=3.9,用甲基橙为指示剂,从黄色滴到橙色,此时NaHCO3 ——完全中和。
误差来源:因为 <1O4,突跃小,酚酞变色不明显,所以V1不可靠。
<1O4,突跃小,酚酞变色不明显,所以V1不可靠。
若不标定盐酸浓度,则混合碱的百分含量不会改变,可不能得到其组分的准确含量。
6. 盐酸溶液为什么用碳酸钠标定?为什么用甲基橙指示剂?写出此滴定反应的方程式。
答:因为碳酸钠的组成与其化学式完全相符,纯度足够高,在通常条件下很稳定,参加反应时按反应式定量进行,没有副反应,所以用其做基准物标定盐酸浓度。
因为甲基橙变色范围为3.1-4.4,此时碳酸钠完全反应,生成二氧化碳和氯化钠,终点变色敏锐,并容易控制,突跃明显。
Na2CO3 + HCl = NaHCO3 + NaCl
NaHCO3 + HCl = NaCl + H2O + CO2
7. 有人在实验过程中称取Na2CO3固体0.1534克,用待标定的浓度约为0.1摩尔/升的盐酸滴定,甲基橙为指示剂,滴加已超过45毫升也不变色,请你分析其中的原因。
答:可能的原因有:溶解碳酸钠过程中加水太多,过于稀释,使终点变色不明显,甚至观察不到;碳酸钠称量不准确;未加指示剂;指示剂失效;称取的碳酸钠不纯,混有其它消耗更多HCl的杂质;盐酸浓度过低,(学生配制溶液的过程中,搅拌不均匀或没有搅拌)。荣国用自来水配制盐酸溶液,也可能出现上述情况。
8. 一般实验室为同学们准备实验时,都是把混合碱配成溶液提供给同学们,请分析一下原因。
答:混合碱固体易吸收空气中的水分和二氧化碳,发生潮解和变质,影响实验结果的精密度,配成溶液避免这种影响,并且提供相同的试样,具有更大的可比性。
9. 减量法使用哪些仪器?称量的关键是什么?
答:仪器有称量瓶、电子天平,小纸条。
关键:天平保持水平;不能用手接触称量瓶,要用纸条套住称量瓶和瓶盖柄,用瓶盖轻轻敲击瓶口,,不要撒在容器外;接近要称的量时,要用称量盖轻轻敲瓶口侧面,使试样落入瓶内,盖好瓶盖;每次都要将天平回零。
10. 取两份相同的混合碱溶液,一份以酚酞作指示剂,另一份用甲酚红-百里酚蓝为指示剂滴定到终点,哪一份消耗的盐酸体积多?为什么?
答:第一份消耗的多。因为酚酞由粉红色完全变为无色时,PH=8.0,并且终点不敏锐,很容易过量,甲酚红-百里酚蓝变色时PH=8.3,颜色变化明显(紫色变为粉红色),终点敏锐,容易判断,从PH值的变化可看出,用酚酞滴定时消耗更多的盐酸。
实验六 BaCl2.2H2O中Ba含量的测定
1.为什么要在稀HCl介质中沉淀BaSO4?HCl加入太多有何影响?
答:加HCl是为了防止生成BaCO3,BaHPO4,BaHAsO4沉淀以及Ba(OH)2,同时增加BaSO4在沉淀过程中的溶解度,降低相对过饱和度,有利于沉淀获得较好的晶形。如果太多,会溶解过多的BaSO4,使沉淀不完全。
2.用沉淀理论来解释本实验的沉淀条件。
答:BaSO4 (s) 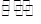 Ba2+ + SO42-
Ba2+ + SO42-
根据溶解平衡,当有过量的SO42-时,平衡向左移动,使Ba2+沉淀完全。因此,本试验中硫酸是相对过量的。
3.试验中沉淀的生成是在热溶液中进行,而过滤又要在冷却后进行,为什么?晶形沉淀为何要陈化?
答:热溶液时反应速率快,加快了沉淀生成的速率。热溶液中的溶解度比冷溶液中大,因此,过滤时,冷却后可以产生更多沉淀,使结果更准确。陈化可以使小晶体长大,利于过滤。
4.为什么要用倾析法过滤?
答:倾析法过滤可以加快过滤速度。
5.灰化的过程中滤纸着火了,应该如何处理?着火对实验结果产生什么影响?
答:滤纸着火应该用坩埚盖盖灭,着火容易产生飞灰,使产物损失。
6.在炭化,灰化的过程中要注意什么?
答:在炭化,灰化过程中,火焰不要太大,应该不包围坩埚。
7.为净化BaSO4沉淀和使之容易过滤,我们在实验中采取了哪些措施?
答:首先陈化晶体,使之长大,洗涤时应注意:滴入洗涤液要轻,防止沉淀溅出;洗涤要少量多次;尽可能使所有洗涤液流完后再进行下一次洗涤。
8.选择沉淀洗涤剂的原则是什么?
答:选择沉淀剂的原则是:使沉淀的溶解度尽量小,不容易引入杂质,或者杂质容易去除。
9.设计一个适合于恒量记录的实验报告格式。
答: 取不同时间对应的重量做表格,直到两次称重相差小于0.0002g时,认为符合要求。
10.坩埚的准备工作该进行哪些操作?
答:恒量坩埚的方法是在马弗炉中800度恒温30~40分钟,放入干燥器中完全冷却后称量。
11.为什么称量0.4~0.6g BaCl2.2H2O?
答:0.4~0.6gBaCl2.2H2O理论上得到的BaSO4为0.38~0.57g。
实验七 纯水的制备与检验水的总硬度测定
1. 自来水中的主要无机杂质是什么?为何蒸馏法和离子交换法能去除水中的无机杂质?新制的蒸馏水和敞口久放的蒸馏水有何差异?
答:自来水中的主要无机杂质有Na+,Ca2+, Mg2+, HCO32-, CO32-, SO42-,S2O32-, Cl-等。
蒸馏法通过加热使含盐的水蒸发,蒸气冷凝成为蒸馏水。而溶解在水中的盐类则残留在蒸馏器具中。离子交换法是先用H+交换水中的阳离子,再将其通过阴离子交换器,用OH-离、交换水中的阴离子,将其混合去除水中的无机杂质。
敞口久的蒸馏水会吸收空气中的CO2等,因而其电导率会上升,而新制的蒸馏水所含的无机杂质较少。
2. 蒸馏装置的搭建的原则是什么?一定是“从左到右”吗?
答:蒸馏装置搭建原则:从左至右,由下至上,横平竖直,拆时反向操作。不一定是“从左到右”,要根据实验环境而定。
3. 为什么加入沸石?如果没有沸石用其他东西可以代替吗?
答:加入沸石,防止过热迸沸。没有沸石可用玻璃珠,毛细管或碎瓷片代替。
4. 为什么要去掉最初的10cm3的馏分?
答:最初的馏分里含有蒸馏装置中的杂质及一些气体杂质。
5. 加入高锰酸钾的目的是什么?
答:除去水中溶有的有机物。
6. 根据蒸馏原理,试回答怎样制取无氨蒸馏水和不含有机物的蒸馏水?
答:制取无氨蒸馏水需要加入PO43-,制取不含有机物蒸馏水需加KMnO4。
7. 测定水的总硬度时,EDTA的标定可采用两种两种方法:①用纯金属锌为基准物质,在pH为5时,以二甲酚橙为指示剂进行标定;②用CaCO3作基准物质,以铬黑T为指示剂,在pH为10时进行标定。请问本实验用哪种标定方法更合理?为什么?
答:第二种标定方法更合理。因为实验后续操作中用EDTA滴定Ca2+。因而为了前后一致,第二种方案更合理。
8. 用含CaCO3的浓度表示水的总硬度,“该数值表明水中CaCO3的真实含量”这种说法对吗?
答:不对。水的总硬度是Ca2+, Mg2+的总含量换算成了CaCO3的质量而表示的,并非CaCO3的真实含量。
9. 取自来水样和蒸馏水后残余水样要注意什么?为什么?
答:取自来水样时,要先取一大烧杯自来水而后分次量取,为的是保证每一次量取的自来水都有相同的状态。
实验八
实验九
实验十(有机)
实验十一(有机)
实验十二(有机)
实验十三 酸碱平衡和沉淀平衡
1. HAc体系,甲基红、溴酚蓝等
NH3H2O体系,可以采用Ca指示剂。
2. 两者都属于两性物质,根据公式计算,两种物质的溶液的H+离子浓度为:

也可以解释为前者只有一个氢根离子,电离常数很小,很难电离,而只是发生与水的水解平衡,生成氢氧根离子,从而使溶液显碱性;后者中含有两个氢根离子,一级电离常数较大,可以电离生成部分氢离子,使溶液显酸性。
3. 配制是指用SbCl3,Bi(NO3)3 ,Na2S的固体配溶液
因 SbCl3+H2O  SbOCl ↓+2HCl
SbOCl ↓+2HCl
加HCl 可抑制水解,故配制时加HCl
Bi(NO3)3+H2O BiNO3 ↓+2HNO3
加HNO3可抑制水解,故配制时加HNO3。
Na2S+H2ONaHS+NaOH
加碱液可抑制水解,故配制时加NaOH。
4.滴定的突跃范围是选择指示剂的依据。应当说最理想的指示剂应该是化学计量点和指示剂的变色点一致,但在实际的分析中不大可能做到。因此,只要选择在滴定突跃范围内发生变化的指示剂,即凡变色点的pH值处于滴定突跃范围内的指示剂均适用,都能使滴定保证足够的准确度(相对误差在0.1%以内)。因此主要是计算pH值。
(1)滴定前 溶液的pH值由HCl的原始浓度决定。
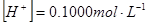 pH=1.00
pH=1.00
(2)滴定开始至化学计量点前 溶液的酸度取决于剩余的盐酸溶液的体积其计算公式为:
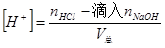
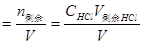
(3)化学计量点时 当滴入NaOH溶液为20.00ml时,到达化学计量点,此时,NaOH和HCl以等物质的量作用溶液呈中性。
 pH=7.00
pH=7.00
(4) 化学计量点后 溶液的pH值取决于过量的NaOH溶液的体积,其计算公式如下:
 =
=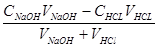
5. 不可以。甲基橙变色范围PH值3.1-4.4,红色变为黄色;而百里酚蓝的变色范围是从6.7到7.5由黄色变为绿色,二者的变色范围相差很大,不可以互换。
实验十四 配合物的生成和性质
1.如何设计实验区分硫酸铁铵和铁氰化钾这两种物质?
答:称取适量的两种物质配成两种溶液,向其中滴加NaOH溶液,出现棕红色沉淀的是硫酸铁铵,无明显现象的是铁氰化钾。
2.在有过量氨存在的[Cu(NH3)4]2+溶液中,加入Na2S、NaOH、HCl,分别对配合物有何影响?
答:[Cu(NH3)]2+Cu2+ + 4NH3
有Na2S存在时,S2-和Cu2+反应生成CuS沉淀,平衡右移,配合物减少;有NaOH时,OH-促使NH3生成,导致平衡左移,配合物增多;有HCl时,HCl和NH3.H2O反应,使平衡右移,配合物减少。
3.写出磺基水杨酸和EDTA的结构,标明配原子的位置。
答:磺基水杨酸
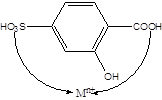 EDTA
EDTA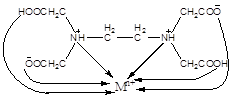
4.从实验设计的角度考虑,平衡移动的实验设计要注意什么才能使实验现象明显?举例说明(用本实验中的内容)。
答:原则是平衡移动,颜色变化明显,这样利于判别。(本实验中在FeCl3溶液中滴加KSCN溶液,然后分散在三个试管中,分别加FeCl3,KSCN,空,可以看到前两个颜色变深,说明平衡向着配合物方向移动。)这个离子不好!
5.Fe3+和C2O42-生成黄色配合物,在此配合物中加入一滴0.1mol.L-1NH4SCN溶液,溶液颜色无明显变化,但向溶液中逐滴加入6mol.L-1HCl溶液后,颜色变红,解释原因。
答:因为开始时Fe3+和C2O42-形成的配合物较稳定,因此配离子中的Fe3+无法与SCN-反应,当加入HCl后,配合物被破坏,Fe3+和SCN-反应生成红色的配合物。
6.设计实验证明[Ag(NH3)2]+配离子的溶液中有Ag+。
答:可以滴加HCl,看是否有白色沉淀生成。
7.如何检验牛奶中有Ca2+?
答:取适量的牛奶于试管中,用碱液调节pH>12,再加少量钙试剂,看是否有红色出现。
实验十五 氧化还原反应和电化学
1. 通常条件下,氧化还原反应总是由较强的氧化剂与还原剂向着生成较弱的氧化剂和还原剂方向进行。从电极电势的数值来看,当氧化剂电对的电势大于还原剂电对的 电势时,反应才可以进行。反应以“高电势的氧化型氧化低电势的还原型”的方向进行。在判断氧化还原反应能否自发进行时,通常指的是正向反应。
任何一个氧化还原反应,原则上都可以设计成原电池。利用原电池的电动势可以判断氧化还原反应进行的方向。由氧化还原反应组成的原电池,在标准状态下,如果 电池的标准电动势 E >0, 则电池反应能自发进行;如果电池的标准电动势 E <0, 则电池反应不能自发进行。在非标准状态下,则用该状态下的电动势来判断。
从原电池的电动势与电极电势之间的关系来看,只有 E > 时,氧化还原反应才能自发地向正反应方向进行。也就是说,氧化剂所在电对的电极电势必须大于还原剂所在电对的电极电势,才能满足E >0的条件。
从热力学讲电池电动势是电池反应进行的推动力。当由氧化还原反应构成的电池的电动势Eφ池大于零时,则此氧化还原反应就能自发进行。因此,电池电动势也是判断氧化还原反应能否进行的判据。
查表知:Fe 3+ + e = Fe2+ j1= +0.771V
Br2 + 2e = 2Br- j2= +1.066V
由反应式可知:Br2 是氧化剂,Fe 是还原剂。
故上述电池反应的 E = +1.066-0.771=0.295V>0。可以正向进行。
2. 向硫酸铜溶液中加入少量氨水,得到的不是氢氧化铜,而是浅蓝色的碱式硫酸铜沉淀:
2CuSO4+2NH3·H2O=(NH3)2SO4+Cu2(OH)2SO4
若继续加入氨水,碱式硫酸铜沉淀就溶解,得到深蓝色的四氨合铜配离子:
Cu2(OH)2SO4+8NH3=2[Cu(NH3)4]2++SO42-+2OH-
3.
4. E3>E7>E4>E5>E1>E2>E6
5.前者均大于后者。生成沉淀和配合物将使电极电势升高。
实验十六 乙酸电离常数和电离度的测定
1. 测溶液pH值是将参比电极和测量电极同时浸入待测溶液中组成电池。25℃时,pH=(E-E0)/0.059。定位就是用已知pH值的标准缓冲溶液定位E,代入E0后,就可以根据其测出未知液E,算出pH值。一般选用接近中性的溶液与饱和KCl溶液作为定位用缓冲溶液。
2. 在测定一组HAc时,电极上会沾有所测剩余溶液,若上一组测较浓溶液,则电极头上会留有大量氢离子,难以除干净,在测下一组HAc时,会对结果产生很大影响。
3. 会有变化。因为电离过程是个吸热过程,升高温度会使平衡反应向正方向移动,即电离反应方向,电离度与电离常数随之升高。降低温度,平衡向反方向移动,电离度与电离常数随之降低。
4. 用去离子水清洗后,再用溶液润洗两到三次。准确量取,读数时要平视液面。若有吹字则吹出最后一滴。
5. 通过实验作图结果与计算结果比较可知作图法精度高。
提高准确度的方法有:1标定HAc浓度时平行滴定三次,求平均值,以减小误差;2 吸取溶液用移液管及吸量管以提高吸取量的准确度;3 实验数据处理时用作图法,使五个作图点尽量多的落在一条直线上或分居此直线的两侧,这样便可平衡各数据的误差。
6. 不能。因为若浓度很稀,水的电离所产生的氢离子就不可以忽略了。而且,平衡时HAc的浓度也将变化,稀释有助于HAc的电离,则其浓度不能用c代替。
7. 因为HAc电离微弱且H2O相对HAc电离更是可以忽略,所以可以认为乙酸根离子与氢离子的浓度相同。通过测定pH值,可知氢离子浓度,即等同于乙酸根离子。HAc电离微弱,可以用初始浓度代替,初始浓度则通过三次平行滴定测出。
8. 可以不烘干。可以用下一组将测的HAc润洗此烧杯,且在电极棒插入溶液前也事先用HAc溶液润洗几次,即可基本消除误差。
实验十七 化学反应速率及活化能的测定
1、 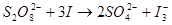 ……(1)
……(1)
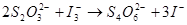 ……(2)
……(2)
反应(2)的速率远大于反应(1)的速率。如果加过硫酸铵的速率不够快,则会出现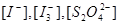 变化不均匀的情况,从而造成反应终点判断不准确,增大实验误差。其次,过硫酸铵易分解。所以必须快速加入过硫酸铵,以减小实验误差。
变化不均匀的情况,从而造成反应终点判断不准确,增大实验误差。其次,过硫酸铵易分解。所以必须快速加入过硫酸铵,以减小实验误差。
2、先计时后震荡,没有使各物料接触均匀,充分就开始计时,则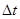 偏大,
偏大,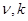 均偏小;而先振荡后计时,则情况恰相反。如果把(NH)2S2O8与Kl的加入顺序对调,则使
均偏小;而先振荡后计时,则情况恰相反。如果把(NH)2S2O8与Kl的加入顺序对调,则使 与
与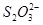 反应,会使实际参与计时反应的减少,而增大实验误差(则偏小,均偏大)。
反应,会使实际参与计时反应的减少,而增大实验误差(则偏小,均偏大)。
3、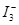 与淀粉溶液显出蓝色,是本实验的计时终点,由本实验中的两个关联方程式(,)知:所加入的Na2S2O3的量决定着的量及其出现的时间,即决定着溶液出现蓝色的时间。
与淀粉溶液显出蓝色,是本实验的计时终点,由本实验中的两个关联方程式(,)知:所加入的Na2S2O3的量决定着的量及其出现的时间,即决定着溶液出现蓝色的时间。
如果加入的Na2S2O3过少,会使计时时间过短,容易造成较大误差;如果加入过多,则反应计时时间内所消耗的(NH)2S2O8过多,近似用平均速率代替瞬时速率的方程就不成立,即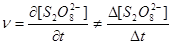 ,甚至会出现(NH)2S2O8消耗完时,还有Na2S2O3剩余,以致无法显示计时终点的情况。
,甚至会出现(NH)2S2O8消耗完时,还有Na2S2O3剩余,以致无法显示计时终点的情况。
本实验中,Cu2+是催化剂,可以降低(NH)2S2O8与Kl反应的活化能,加速反应,且随[Cu2+]的增加,反应加快。
4、可能造成量筒中的残液与所量取液的反应,使所量取液的有效量减少,对结果造成较大误差。
与反应而造成较大误差。
6、所得的数据不同,作图法更合理。因为通过合理选取数据点作图,可以摒弃一些有较大实验误差的数据,并可以综合处理各方面的误差,使得结果更为合理准确。
通过本实验可以总结出影响化学反应速率的因素有3条:反应物浓度,反应体系的温度,催化剂及其用量。
7、求平均值和作图法
十八水杨酸合铜
1,不能,由于等摩尔数连续变化法要求保持总物质的浓度不变,如果有多种配合物,可能产生不能的配比,那么就无法保证这个前提。
2,a,不使用时用及时打开样品室盖子,断开光路,避免光电管老化;b,测试前需要预热15分钟;c,每次改变波长需要重新调零,同时在实验过程中应该随时检查吸光度零点是否有变化并及时做出调整。
3,硝酸钾溶液。
4,因为两种原始溶液——硝酸铜和璜基水杨酸溶液的物质量浓度均为0.05mol/L,所以在这里的计算过程中n之比值相等于V之比值。
5,[CuC7H4O3R]2+, [Cu(C7H4O3R)2]2+
实验十九 铅铋合金中Pb2+、Bi3+含量的连续测定
1. 加入六次甲基四胺的目的是缓冲上一步中过量的盐酸,以调节pH(未加之前pH约为5.25)至合适范围(pH=6左右)。过量的目的是缓冲与EDTA络合过程中产生的氢离子。
2. 不能。pH在5-6之间时,Bi3+已水解。
3. 首先调节pH≈1,滴加二甲酚橙指示剂后,Bi3+与指示剂形成紫红色配合物,这是因为二甲酚橙与金属离子形成的配合物都是红紫色。二甲酚橙与Pb2+、Bi3+形成的络合物呈红色,它们的稳定性小于Pb2+、Bi3+和EDTA所形成的络合物。当EDTA滴加量逐渐增多时,Bi3+与EDTA形成黄色络合物,当溶液由紫红色转变为亮黄色时,即为滴定终点。由于在滴定Bi3+时,加入了较大量的酸,因此在滴定Pb2+时加入过量六次甲基四胺溶液,以缓冲过量的酸,并调节溶液pH至5~6,在此pH 下Pb2+可与二甲酚橙形成紫红色配合物。但此配合物同样不如其与EDTA所形成配合物稳定。当用EDTA标液滴定时,Pb2+与EDTA形成黄色络合物,当溶液由紫红色转变为亮黄色时,即为滴定终点。
4. 可以。由于EDTA本身在水中的溶解度较小,且通常含有少量水和杂质,因此通常用其二钠盐来配制EDTA标液。
5. 氨水的缓冲区为pH 9~10,在本实验的pH范围内不能形成缓冲作用,而碱则只中和酸而不能形成缓冲体系。由于Ac-离子能与Pb2+形成络合物,将会影响Pb2+的准确滴定。
6. 标定EDTA溶液的基准物有,Zn,ZnO,CaCO3,Cu,Bi,MgSO4·7H2O,Ni,Pb等。常见用纯金属锌作基准物标定EDTA,可以用铬黑T作指示剂,用氨缓冲溶液,在pH=10进行标定。也可用二甲酚橙作指示剂,用六亚甲基四胺调节酸度,在pH=5~6进行标定。
实验二十 COD测定
1. 滴定过程应先慢后快再慢,开始时应逐滴滴加,待红色褪去再滴加第二滴;之后,由于反应生成Mn2+,发生自催化作用,因此滴定速度可加快;快到终点时应慢滴以利于终点的把握。
2. 可选用酸式滴定管。吸量管,移液管均可,但由于器材数量限制,也可用酸式滴定管精确量取10.00mL KMnO4。滴定管的精度也是小数点后两位数。
3. 若不加硫酸,则KMnO4可能与自来水中其他杂质离子发生氧化还原反应,使得最后回滴时的结果偏大。
4. 本实验的主要不同在于采用了返滴定的方法。该方法可有效避免直接滴定时终点颜色(由深到浅)不易判断的问题。同时长时间在沸水浴加热可最大限度除去有机杂质,使结果更合理。
5. 返滴定法。若直接用Na2C2O4滴定过量的KMnO4,则终点时溶液颜色由深紫红色变成无色,不易准确判断,不符合终点颜色变化原则。采用返滴定法时,终点颜色由无色变成微红色,较易判断。加热的目的是保证有机物与KMnO4反应完全。
Cl-含量高时,其在酸性中可能被KMnO4氧化。发生如下反应
10Cl- + 2MnO4- +16H+ ==5Cl2 +2Mn2+ +8H2O
使得测定结果偏高。
6. COD可反应水质受有机物或还原性物质污染程度,可作为水中有机物含量的指标之一。测定COD的方法一般有高锰酸钾法,重铬酸钾法和库仑法。
7.
二十一硫酸铵中氮含量测定
1, 首先是甲醛中微量酸的影响,如果不事先中和,会造成最终结果偏大;第二是试样中游离酸的存在,使得结果偏大。
2, 除了硝酸铵外,其他都是铵态氮可以用此法测定,而硝酸铵中硝酸根与铵根是1:1的关系,同时硝酸根不参与反应,因此也可以用此法测定总氮含量。
3, 这些离子均能参与反应,与氢氧化钠标准液反应能使结果偏高。
4, 是根据指示剂甲基红的指示范围为4.4-6.2,可以防止体系过于碱性,减小误差。
5, 第一种方法较精确,但是终点指示模糊,容易在此产生误差。第二种方法易于观察,但由于酚酞的指示范围在8.0-9.6,实际上滴定游离酸所用的NaOH相对于另一份试液已经过多,从而会造成实验结果偏大。
6, 可以不事先处理甲醛,其他过程与本实验相同,对比实验结果的误差,如果在误差允许的范围内,则说明甲醛中游离酸对实验结果的影响可以忽略不计。
7, 酚酞指示范围为8.0-9.6,是最常用的碱滴定酸的指示剂,一方面变色范围比较准确,一方面变色点十分容易观察。
实验二十二 非水滴定法
1 非水滴定已有五十多年的发展历史。1956年在理论上取得了重大突破,近二十年来取得了长足的发展。
非水滴定是指用水以外的其它溶剂作为滴定介质的一种容量分析法。根据酸碱的质子理论,物质的酸碱性除了于物质的本质有关外,还与溶剂的性质有关。溶剂的固有酸碱度能影响溶质的表观酸碱强度。溶剂的酸度越强,溶质的表观酸度就越弱,而其表观碱度则增强。反之亦然。这种影响的大小取决于溶质与溶剂的反应程度。
非水溶剂对有机化合物的溶解能力比水强,因此适用于分子量大,极性弱的有机酸、有机碱、高级脂肪酸等。另外,本身与水反应的某些物质可以用适宜的非水滴定体系。
常见的非水滴定体系有:乙酸、盐酸—丙酮、溴化氢—冰醋酸等
2、温度、实验器具、滴定速度、终点判断
3、铵盐,如NH4Cl,NH4NO3,(NH4)2C2O4
实验二十三 沉淀滴定法测定调味品中氯化钠的含量
1、 去除其中的水分
会使所标定的AgNO3溶液浓度偏小。
2、加硝酸是除去一些弱酸根离子的影响,如:CO、Ac,……。再加KMnO4,是因为酸性条件下,KMnO4的氧化性较强,可以去除还原性物质对AgNO3的影响
3、用铁铵矾作为指示剂的银量法称为福尔哈德法。
因为要使用AgNO3(原因同上2),且测定调味品(酱油、味精)中含有弱酸和还原性物质。
4、同2
5、不需要。
因为比较三者的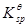 ,AgBr(5.0*10-13)和AgI(8.5*10-17)比AgSCN(1.0*10-12)更小,沉淀不会转化。
,AgBr(5.0*10-13)和AgI(8.5*10-17)比AgSCN(1.0*10-12)更小,沉淀不会转化。
6、应使用王水,应为试管壁上可能会附着有被还原的Ag,王水能溶解。
7、沉淀中会附着银离子,从而使NHSCN的用量减少,而总结果偏小,加入一定的有机试剂的覆盖法,或另外选一种配位滴定法会更好
实验二十四 胃舒平药片中铝、镁的测定
1. 实验中为什么要称取大量样品溶解后再分取部分试样进行实验?
答:因为胃舒平药片中各组分含量可能不均匀,为使测定结果具有代表性,本实验取较多样品,研细混合后再取部分进行分析。
2. 在分离Al3+后的滤液中测定Mg2+,为什么还要加入三乙醇胺溶液?
答:加入三乙醇胺的作用是掩蔽滤液中仍可能存在的Al3+。
3.测定Mg2+时可否不用分离Al3+,而是采用掩蔽的方法直接测定Mg2+?根据Al3+的性质选择什么物质掩蔽比较好?设计实验方案。
答:可以。可采用DDTC、盐酸羟胺、三乙醇胺联合掩蔽剂掩蔽大量的铝,选择合适的掩蔽剂用量后先用掩蔽剂用量,加入掩蔽剂掩蔽Al3+后,在pH=l0条件下,以铬黑T为指示剂,用EDTA滴至溶液由紫红色变为纯蓝色为终点。
4. 能否改进实验条件用EDTA标准溶液直接滴定Al3+?
答:不能。因为Al3+的滴定存在下列问题,Al3+对二甲酚橙等指示剂有封闭作用;Al3+与EDTA配位缓慢,需要加过量EDTA并加热煮沸,配位反应才比较完全;在酸度不高时,Al3+水解生成一系列多核氢氧基配合物,如[Al2(H2O)6(OH)3]3+,[Al3(H2O)6(OH)6]3+等,即便将酸度提高至EDTA滴定Al3+的最高酸度(pH=4.1),仍不能避免多核配合物的形成。铝的多核配合物与EDTA反应缓慢,配位比不恒定,故对滴定不利。故不宜采用直接滴定法,应采用返滴定法。
5. 在滴定Al3+和Mg2+之前,为什么要加入氨水产生沉淀,并用盐酸将沉淀溶解?解释这一步骤的目的和机理。
答:滴加氨水是为了将样品处理中所用的过量的盐酸中和,当刚好产生沉淀时刚好中和,再用盐酸中和稍过量的氨水,沉淀刚好溶解时溶液刚好呈中性。
6. 在滴定Al3+和Mg2+时选用不同的pH,解释选择的依据。
答:滴定Al3+时调节PH到4左右是为了防止Al3+水解。而滴定Mg2+时由于MgY2-(lgKMgY=8.7)稳定性比较差,根据pH-lgKMY曲线,必须在弱碱性pH=10的条件下滴定,且指示剂铬黑T的pH范围为7~10。
7. 该实验中对Al3+和Mg2+的测定可否会受到其他离子如Ca2+、Zn2+、Mn2+、Fe3+的干扰?
答:由如图所示EDTA的酸效应曲线可以看出,Mg2+与Ca2+、Mn2+的稳定常数较接近,因此受其干扰较大,而Al3+与Ca2+、Zn2+、Mn2+、Fe3+的稳定常数都较接近而且允许的pH值也较接近,故受Ca2+、Zn2+、Mn2+、Fe3+离子的干扰都较大。

EDTA酸效应曲线
第十章(有机)
实验三十五 非金属元素性质综合实验
1. 实验室的H2S、Na2S、Na2SO3溶液能否长期保存?说明理由。
答:不能。因为三者均易被空气中的O2氧化而变质。
2. 选用一种试剂区别下列五种无色溶液:NaCl,NaNO3,Na2S,Na2S2O3,Na2HPO4。
答:选用AgNO3:
Cl- + Ag+ = AgCl↓
NaNO3与AgNO3不反应。
S2- + 2Ag+ = Ag2S↓(黑)
S2O32- + 2Ag+ = Ag2S2O3↓(白) Ag2S2O3 + H2O = Ag2S↓(黑) + H2SO4
HPO42- = H+ + PO43- PO43- + 3Ag+ = Ag3PO4↓(黄)
3. 在选用酸溶液作为氧化还原反应的介质时,为何不常用HNO3或HCl?在什么情况下可选用HNO3或HCl?
答:因为HNO3具有氧化性,而HCl具有还原性,会干扰氧化还原反应的进行。当HNO3或HCl不会参与氧化还原反应时,才可以作为氧化还原反应的介质使用。
4. 有三瓶未贴标签的溶液分别是NaNO2、Na2SO3和KI,如何进行鉴别?
答:分别取少量三种试液,加入Cl2和CCl4,CCl4层呈紫红色的试液为KI;再向另外两只试管中加入少量BaSO4,出现白色沉淀的试液为Na2SO3,另外一种试液即为NaNO2。相关方程式如下:
Cl2 + 2NO2- = 2NO3- + 2Cl-
Cl2 + 2I- = I2 + 2Cl-
Cl2 + SO32- = SO42- + 2Cl-
SO42- + Ba2+ = BaSO4↓
5. KIO3和NaHSO3在酸性介质中的反应产物与二者的相对量有什么关系?
答:2IO3- + 5HSO3- = I2 + 5SO42- + H2O + 3H+
I2 + HSO3- + H2O = 2I- + SO42- + 3H+
当nKIO3 : nNaHSO3≥2 : 5时,生成I2、SO42-;
当nKIO3 : nNaHSO3<1 : 3时,生成I- 、SO42-;
当1 : 3<nKIO3 : nNaHSO3≤2 : 5时,生成I2、I-、SO42-;
实验三十六 金属元素性质综合实验
1. 在Cr3+溶液中加入Na2S,得到的沉淀是什么?解释之。
答:Cr(OH)3。因为Na2S易发生水解反应。
2. 某同学在鉴定Cr3+时,向Cr3+溶液中加入NaOH并用H2O2氧化,然后加入稀H2SO4、乙醚,却得不到蓝色而是绿色的溶液,请你帮助分析原因。
答:原因:①在加入H2O2后,应该用水浴加热;②应该用浓H2SO4,而不是稀H2SO4。
3. Cu(Ⅰ)和Cu(Ⅱ)各自稳定存在和相互转化的条件是什么?
答:当Cu(Ⅰ)以配合物或难溶物形式存在时,可以稳定存在;而Cu(Ⅱ)在酸性溶液中可以稳定存在。在Cu(Ⅱ)溶液中加入还原剂如Cu粉时,可以转化为Cu(Ⅰ);而Cu(Ⅰ)在溶液中极不稳定,容易发生歧化反应,生成Cu和Cu2+。
4.如何制得白色的Mn(OH)2沉淀?
答:在试管中加入少量MnSO4溶液,加入适量密度小于水的有机溶剂如甲苯、乙醚等进行液封,然后加入少量的NaOH溶液,在下层水相就会出现白色的Mn(OH)2沉淀。
5. 碱性介质中Co(Ⅱ)能被Cl2氧化Co(Ⅲ),而酸性介质中Co(Ⅲ)又能将Cl-氧化为Cl2,Co(Ⅱ)的还原性弱而[Co(NH3)6]2+又极易被空气氧化氧化为[Co(NH3)6]3+,如何解释这些“矛盾”的现象?
答:在碱性介质中,ФΘCo(Ⅱ) /Co(Ⅲ)<ФΘCl2/Cl-,发生氧化还原反应时,Cl2作为氧化剂可以将Co(Ⅱ)氧化成Co(Ⅲ);而在酸性介质中,ФΘCo(Ⅱ) /Co(Ⅲ)>ФΘCl2/Cl-,发生氧化还原反应时,Co(Ⅲ)将Cl-氧化为Cl2。
[Co(NH3)6]3+的稳定性好于[Co(NH3)6]2+,是因为[Co(NH3)6]3+形成的是内轨型配合物,而[Co(NH3)6]2+形成的是外轨型配合物,[Co(NH3)6]3+稳定容易生成,所以即使Co(Ⅱ)的还原性较弱, [Co(NH3)6]2+也极易被空气氧化氧化为[Co(NH3)6]3+。
实验三十七 金属元素综合设计性实验
实验三十八(有机)
实验三十九 硫酸亚铁铵的制备和硫酸亚铁质量分数的测定(题目加上)
Ⅰ 硫酸亚铁铵的制备
1. 可接上盛有CuSO4溶液的洗气瓶,吸收产生的有毒气体,反应式如下
Cu2+ +H2S==CuS +2H+
8CuSO4 +PH3 +4H2O==4CuSO4 +4H2SO4 +H3PO4
3CuSO4 +2PH3==3H2SO4+2Cu3P
4CuSO4 +PH3 +4H2O==H3PO4 +4H2SO4 +8Cu↓
也可以用酸性KMnO4溶液除去。(写上反应方程式)
2. 加入少量铁屑是为了防止Fe2+被氧化成Fe3+:
Fe3+ + Fe==Fe2+
3. Fe2+在酸性溶液中稳定存在,因此加入H2SO4保持溶液的酸性,防止Fe2+被氧化或水解。加入少量蒸馏水是使溶液浓度降低,防止过滤时析出晶体。
4. FeSO4溶解度随温度变化较大,温度较高时溶解度较大,因此为了避免过滤时析出过多晶体,需趁热过滤。绿色晶体应该是FeSO4水合晶体,可能是在过滤时,由于漏斗柱温度较低,热的FeSO4溶液流经漏斗柱时发生热交换而降低温度,析出晶体。用少量不含氧气的蒸馏水淋洗溶解即可。(加一个溶解度曲线图,根据书上的数据)
5. 保持酸度是为了防止Fe2+被氧化成Fe3+,同时防止其水解。
6. 这是由于FeSO4的溶解度受温度影响较大,当热的FeSO4溶液遇到温度较低的漏斗时,发生热交换,溶液温度降低,析出FeSO4水合晶体而堵塞滤纸孔,造成过滤速度较慢。(如何处理?)
7. (NH4)2FeSO4·6H2O在乙醇中溶解度很小,但在水中溶解度较大,因此不能用蒸馏水淋洗。
8. O2可氧化Fe2+变成Fe3+,从而使测定结果偏高。限量分析反应式为:
Fe3+ +n SCN- ==[Fe(NCS)n]3-n
8. 所用仪器有:吸滤瓶,布氏漏斗,抽滤装置;
注意事项:安装布氏漏斗时,应把布氏漏斗下端的斜口与吸滤瓶支管相对;吸滤用的滤纸应比布氏漏斗的内径略小,以恰好盖住瓷板上所有的孔为度;放好滤纸后,先用少量去离子水润湿,再打开开关,布氏漏斗中的液体不得超过漏斗溶剂的三分之二;停止吸滤时,应先拆下吸滤瓶上的橡皮管或拔去漏斗,然后关闭抽滤装置,以防止水倒吸。
10. 应以铁为准。加入的H2SO4是过量的,同时(NH4)2SO4晶体的理论计算量是以铁为基准的。
11.?
12. (NH4)2FeSO4·6H2O和NaCl的热稳定性不同,含结晶水的情况不同。溶解度受温度影响也不同。(NH4)2FeSO4·6H2O热稳定性较差,若加热过度,二价铁可能被氧化,NaCl的热稳定性较好,加热至黏稠状影响不大。(NH4)2FeSO4·6H2O的溶解度受温度影响较大,因此采用冷却热饱和溶液制得晶体,而NaCl的溶解度受温度影响较小,因此可蒸发溶剂至溶液呈黏稠状。
Ⅱ 硫酸亚铁铵中的Fe2+含量的测定
Fe含量的测定方法有:邻二氮菲分光光度计法;重铬酸钾法;
邻二氮菲分光光度计法可参照前面实验:邻二氮菲吸光光度法测定铁含量;
重铬酸钾法测定铁的实验方案:
1. 配制K2Cr2O7标准溶液
准确称取固体0.9~1.1g置于250ml烧杯,加50ml水溶解,定容200ml容量瓶中,摇匀。
2. 亚铁盐中铁含量的测定
(1)准确称取0.8~1.2g试样3份于锥形瓶中,加50ml水,10ml硫酸,5ml磷酸,加6d二苯胺磺酸钠;
(2)用K2Cr2O7标准溶液滴定至溶液由绿色突变为紫色或紫蓝色为终点;
(3)计算浓度。
3. 可用NH4F作掩蔽剂,形成稳定的[FeF6]3-络离子;或加入少量5%盐酸羟胺溶液以消除其干扰。
实验四十 三乙二酸合铁(Ⅲ)酸钾的制备
1. 在由黄色沉淀制备绿色化合物时,加入了H2O2溶液,有红棕色沉淀生成,用方程式表示这一制备过程。
答: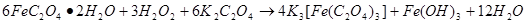
2. 实验过程中使用的氧化剂为H2O2,仔细观察实验现象,据此写出H2O2发生的反应方程式。
答:
3. 在这个实验中,最后一步能否用蒸干溶液的方法来提高产率,为什么?
答:因为K3[Fe(C2O4)3]·3H2O在110℃下可失去部分结晶水,230℃时分解。
4. 在最后的溶液中,加入乙醇的作用是什么?悬挂棉线的作用是什么?
答:乙醇起到溶剂的重结晶作用。利用溶剂对被提纯物质及杂质的溶解度不同,可以使被提纯物质从过饱和溶液中析出。而让杂质全部或大部分仍留在溶液中(若在溶剂中的溶解度极小,则配成饱和溶液后被过滤除去),从而达到提纯目的。 悬挂棉线的作用是晶体可围绕棉线生长。
5. 加入H2O2后为何要再加入饱和H2C2O4?然后为什么要趁热过滤?
答:加入适量的乙二酸可使Fe(OH)3转化为三乙二酸合铁(Ⅲ)酸钾配合物,方程式为:
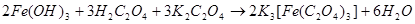 。趁热过滤可滤去杂质,以免在晶体中混入杂质,并且可提高产率。
。趁热过滤可滤去杂质,以免在晶体中混入杂质,并且可提高产率。
6. 如何确定K3[Fe(C2O4)3]·3H2O的组成?简单说明。
答:检定K+:取少量K2C2O4及产品溶液,分别与饱和酒石酸氢钠(NaHC4H4O6)溶液作用。充分摇匀,观察现象是否相同;检定C2O42-:在少量K2C2O4及产品溶液中分别加2滴CaCl2溶液,观察现象有何不同;检定Fe3+:在少量FeCl3及产品溶液中,分别加入1滴KSCN溶液,观察现象有何不同。
7. 合成过程中,为何第一步生成乙二酸亚铁时加入饱和乙二酸,而在第二步合成三乙二酸合铁(Ⅲ)酸钾时却加入饱和乙二酸钾?试说明原因。
答:第一步时乙二酸亚铁易被氧化成三价铁,故应在酸性条件下反应,所以加入乙二酸,而第二步合成三乙二酸合铁(Ⅲ)酸钾本身主三价铁不会被氧化,可以用盐。
四十一废铝
1, 硫酸在常温下会使铝片表面钝化,从而难以溶解,所以在加热情况下才能溶解。氢氧化钠可以在常温下溶解铝片,但反应过于剧烈。
2, 6.4克硫酸铝。3.3克硫酸钾,生成8.8克明矾。
3, 先用氢氧化钠溶解铝,然后过滤去除铁,然后再加酸调节pH。
4, 用纯水反复洗涤,抽滤。
四十二甘氨酸合铜
1, 顺式甘氨酸铜空间位阻相对较小,更易于与水中氢离子结合,所以溶解度大。这两种异构体的结晶结构和颜色不同,顺式为蓝色针状晶体,反式为蓝紫色鳞片状晶体。
2, 不能,因为直接加入氢氧化钠制得的氢氧化铜为沉淀,无法参与下一步反应,而用氨水处理过后反应生成的是氢氧化铜胶体,胶体与反应物接触面积大,从而能够发生下一步反应。
3, 略
4, 因为在刚抽滤时体系中还有其他化合物存在,直接用乙醇洗涤很可能导致这些化合物析出依附于晶体上。
5, 碘量法测定铜,EDTA-甲醛法测定铜。
由于碘化铜沉淀表面会吸附一些碘使滴定终点不明显,影响精确度,所以在化学计量点时加入少量硫氰化铵,使碘化铜沉淀转变为硫氰化铜,而硫氰化铜比碘化铜溶解度小得多,因此能使被吸附的碘从沉淀表面置换出来。
实验四十三 硫代硫酸钠制备,性质监测与含量测定(不全,请你自己做好)
1.
2.
3.
4.
5.硫代硫酸钠溶液滴定铜,锥形瓶中的溶液达到终点,放置几分钟后慢慢变蓝,分析原因。
答:在硫代硫酸钠溶液滴定铜时,首先铜与过量碘化钾作用析出I2,I2用硫代硫酸钠标准溶液滴定后产生I-,放置几分钟后I-又会被空气中的氧化产生I2,I2遇淀粉批示剂变蓝。
6.推导出计算Na2S2O3含量的公式。
答:假设称取Na2S2O3·5H2O产品的质量为mT,配制成250ml溶液。称取Cu的质量为mCu,所用去溶液的体积为V,则根据Cu2+与S2O32-反应一比一的计量关系,可推导出产品中Na2S2O3·5H2O的质量为: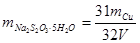 。故Na2S2O3·5H2O含量为
。故Na2S2O3·5H2O含量为 。
。
实验四十四 杂多酸的合成,表征和催化活性的研究
1、 酯化率=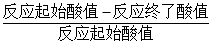
影响醇酸反应酯化率的主要因素有:反应的醇/酸摩尔比;反应的温度、压力、时间;催化剂及其用量;带水剂;反应器
2、杂多酸的催化活性与其杂多阴离子的软度(softness),酸强度,负载剂,催化剂浓度及溶剂密切相关
杂多酸在化学催化剂领域中的应用主要有:烯烃的水合反应,酯分解、酯交换和酯化反应。此外,在化学合成,抗病毒和材料学领域也有其独特的应用。
实验四十五 纳米ZnO的制备和质量分析
(题目加上)
1、用小量筒测定其堆积体积,然后称量计算出密度。这种方法误差较大。
2、制备纳米微粒的方法大体可分为气相法,液相法和固相法。而这三种方法中又有具体细分。
气相法中包含:电阻加热法、高频感应加热法、等离子体加热法、电子束加热法、激光加热法、通电加热蒸发法、流动油面上真空沉积法、爆炸丝法、热管炉加热化学气相反应法、激光诱导气相反应法、等离子加强化学气相反应法、化学气相凝聚溅射法。
液相法:共沉淀法、均相沉淀法、无机盐水解法、金属醇盐水解法、喷雾干燥法、雾化水解法、喷雾培烧法、高温高压水热法、高温高压有机溶剂热法、蒸发溶剂热解法、常压氧化还原水溶液法、常压氧化还原有机溶液法、乳液法、溅射化学合成法、溶胶-凝胶法。
固相法:热分解法、固相反应法、电火花放电法、溶出法、球磨法。
3、制备纳米ZnO材料的方法按物质的原始状态分为固相法、液相法、气相法3类,其中液相法具有过程简单、工业化成本低、产物组成易控等优点,应用尤为广泛,如溶胶凝胶法 、水热法、喷雾热解法、沉积法、微乳液法、模板合成法、分子束外延、磁控溅射等,
水热法具有反应速度快、产物性能优越等特点,日益引起关注
溶胶- 凝胶法因其制备均匀度高、纯度高及反应温度低、易于控制等优点,吸引了诸多的关注. 通常,溶胶- 凝胶法利用金属醇盐(烷氧基金属)为先驱物,通过它们的水解和缩聚进行反应,最终合成纳米级ZnO粉体,但是这种方法需首先制备锌醇盐,制备工艺复杂,成本高
直接沉淀法是液相法中最简单可行的一种,也是目前国内外制备纳米氧化锌的主要方法。液相法是通过液相合成粉料,由于组分分散在液相中,各组分的含量可精确控制,并可实现分子/原子水平上的均匀混合,通过工艺条件的精确控制,可使生成的粉料晶粒尺寸小、粒度分布窄
4、本实验中的两种前躯体更好。首先,氢氧化锌的Ksp远小于碳酸锌,锌转化的更完全。再者,氢氧化锌更易得。
实验四十六 三氯化六氨合钴(Ⅱ)的制备及组成测定
1. 钴(Ⅱ)的热稳定性大于钴(Ⅲ),钴在简单化合物中以钴(Ⅱ)最稳定,钴(Ⅲ)离子是强氧化剂,容易被还原。在酸性介质中,二价钴盐比三价钴盐稳定,而在其配合物中,大多数三价钴的配合物比二价钴的配合物稳定。
2.
3. 三氯化六氨合钴(III)煮沸时可被强碱所分解,放出氨来(反应式如下)。
2[Co(NH3)6]Cl3 + 6NaOH == 2Co(OH)3 + 12NH3 + 6NaCl
逸出的氨用过量的标准HCl 溶液吸收,剩余的酸用标准NaOH 溶液回滴,便可测定出氨的含量。Cl-的含量沉淀滴定法测定(反应式如下)。
Ag++Cl-==AgCl↓
4. 黑色沉淀为氢氧化钴被氧化后产生的三氧化二钴水合物Cr2O3·xH2O。
5.
6. 不可加热至沸。因为生成的三价钴离子可能水解生成氢氧化钴,并可进一步被氧化生成三氧化二钴水合物。
7. 慢慢滴加H2O2是为了防止其分解,同时避免局部浓度过大,造成氧化反应不均。慢慢滴加HCl也是为了防止局部浓度过大,导致无法完全析出晶体。H2O2的作用是将二价钴氧化成三价钴。HCl的作用是中和过量的氨水。
8. ?
实验四十七 (有机)
实验四十八 (你做吧)
实验四十九 (有机)
实验五十 (有机)
实验五十一(有机)
实验五十一 (有机)
实验五十二(有机)
-
《分析化学实验》总结
定量分析是一门实验性课程,通过分析化学实验可以是我们对分析学化学基本理论进一步加深理解,并能够熟练地掌握定量分析的基本操作技能,为…
-
无机与分析化学实验总结
无机与分析化学实验总结一个学期的化学实验了,通过十几个化学实验,我收获很多。我最喜欢做的是结晶的试验,看着从溶液中析出的各种形状的…
-
分析化学实验心得
分析化学实验心得体会分析化学是人们获得物质化学组成和结构信息的科学,它所要解决的问题是物质中含有哪些组分,各个组分的含量多少,以及…
-
分析化学实验心得
Subject:Body-SiteVariationofSkinAutofluorescenceB89203004化學二儀器/實驗…
-
分析化学实验学习感受
《分析化学实验》总结分析化学实验课暂时告一段落,它虽然课时不多,但在这三次实验中,我受益匪浅。《分析化学实验》这门课程的开设是十分…
-
常见物质的分离、提纯和鉴别方法总结
金太阳新课标资源网常见物质的分离提纯和鉴别方法总结一物质的分离与提纯方法1混合物的物理分离方法2混合物的化学分离法第1页共13页金…
-
南京农业大学有机化学考研实验题第四章
第四章天然产物的提取天然产物种类繁多广泛存在于自然界中多数天然产物的提取物具有特殊的生理效能可用作药物香料和染料天然产物的分离提纯…
-
物质的鉴别推断与提纯知识总结
物质的鉴别一初中化学常见物质的颜色二初中化学敞口置于空气中质量改变的一质量增加的1由于吸水而增加的氢氧化钠固体氯化钙浓硫酸2由于跟…
-
总结〗化学方法提纯和分离物质的“四原则”和“三必须”
必修1高一化学教学案归纳几种分离和提纯方法2三必须是一除杂试剂必须过量二过量试剂必须除尽因为过量试剂带入新的杂质三除杂途径选最佳例…
-
26物质的分离与提纯
26物质的分离和提纯教学目标知识技能复习常见物质分离和提纯的实验知识掌握常见物质分离提纯的一般方法能力培养通过对常见物质分离与提纯…
-
无机与分析化学实验总结
无机与分析化学实验总结一个学期的化学实验了,通过十几个化学实验,我收获很多。我最喜欢做的是结晶的试验,看着从溶液中析出的各种形状的…