氧化还原滴定法和配位滴定法实验自修报告 2
氧化还原滴定法
一、概述
氧化还原滴定法是以氧化还原反应为基础的滴定分析方法。
氧化还原滴定法在药物分析中应用广泛,用于测定具有氧化性和还原性的物质,对不具有氧化性或还原性的物质,可进行间接测定。
氧化还原反应较复杂,常伴有各种副反应,反应速度较慢,因此,氧化还原滴定法要注意选择合适条件使反应能定量、迅速、完全进行.
反应条件:
1. 滴定反应必须按一定的化学反应式定量反应,且反应完全,无副反应。
2. 反应速度必须足够快。
3. 必须有适当的方法确定化学计量点
氧化还原滴定法终点的判断
(一)自身指示剂
如 KMnO4 滴定H2C2O4时, KMnO4 既是标准溶液又是指示剂。
(二)特殊指示剂
如用于碘量法中的淀粉溶液,本身不参与氧化还原反应,但它能与氧化剂作用产生特殊的颜色,因而可指示终点。
(三)氧化还原指示剂
二、高锰酸钾法
(一)基本原理和条件
高锰酸钾法是以具有强氧化能力的高锰酸钾做标准溶液,利用其氧化还原滴定原理来测定其他物质的滴定分析方法。强酸性溶液中:
MnO4- + 8H + + 5e- → Mn2- + 4H2O
酸度太高时,会导致高锰酸钾分解,因此酸度控制常用3mol/L的H2SO4来调节,但不能用HNO3或HCl来控制酸度。因为硝酸具有氧化性会与被测物反应,而盐酸具有还原性能与KMnO4反应。
(二)测定方法
1.直接滴定法
由于高锰酸钾氧化能力强,滴定时无需另加指示剂,可直接滴定具有还原性的物质。
2.返滴定法
可测定一些不能直接滴定的氧化性和还原性物质。
3.间接滴定法
有些非氧化性或还原性物质不能用直接滴定法或返滴定法测定时,可采用此法。
(三)应用(市售过氧化氢中H2O2含量的测定)
1.高锰酸钾溶液的配制
市售KMnO4 试剂常含有杂质,而且在光、热等条件下不稳定,会分解变质。因此高锰酸钾标准溶液不能直接配制使用,通常先配成浓溶液放置储存,需要时再取适量稀释成近似浓度的溶液,然后标定使用。
在台秤上称取0.86克固体KMnO4,置于500mL烧杯中,用新煮沸过的冷蒸馏水分数次充分搅拌溶解,置于棕色试剂瓶中,稀释至500mL,摇匀,塞紧,放在暗处静置7~10天(或溶于蒸馏水后加热煮沸10~20分钟,放置2天),然后用烧结玻璃漏斗过滤,存入另一洁净的棕色瓶中储存备用。
2、高锰酸钾溶液的标定
可用基准物质草酸钠标定高锰酸钾溶液的浓度,反应式如下:
2MnO4-+5C2O42- +16H+ =10CO2+Mn2+ + 8H2O
3.过氧化氢含量的测定
过氧化氢具有还原性,在酸性个质中和室温条件下能被高锰酸钾定量氧化,其反应方程式为:
2MnO4- + 5H2O2 + 6H+ = 2Mn2+ + 5O2↑+ 8H2O
测定过氧化氢时,可用高锰酸钾作滴定剂,微过量的高锰酸钾为紫红色显示终点。根据高锰酸钾的浓度和滴定所耗用的体积可以计算出溶液中过氧化氢的含量。
注意事项:
1、标定KMnO4溶液时要注意“三度一点”,分别是:温度、酸度、滴定速度和滴定终点。
2、KMnO4 与H2O2的反应在滴定开始时反应较慢,随着Mn2+生成而加速,可先加入少量Mn2+为催化剂。
3、准确读取滴定管中高锰酸钾溶液的液面读数。
4、测定过氧化氢时要注意催化剂的加入及终点颜色的变化。
5、H2O2具有强氧化性,对环境无污染,使用时避免接触皮肤。
6、H2O2受热易分解,滴定时不需加热。
三、碘量法
(一)基本原理
碘量法是利用碘的氧化性、碘离子的还原性进行物质含量测定的方法。I2是较弱的氧化剂;I-是中等强度的还原剂。
碘量法可用直接测定和间接测定两种方式进行。
1、直接碘量法(或碘滴定法)
l 直接碘量法是直接用I2标准溶液滴定还原性物质,又叫做碘滴定法。
l 直接碘量法只能在酸性、中性或弱碱性溶液中进行。
l 直接碘量法可用淀粉指示剂指示终点。
l 直接碘量法还可利用碘自身的颜色指示终点,化学计量点后,溶液中稍过量的碘显黄色而指示终点。
2、间接碘量法(或滴定碘法)
对氧化性物质,可在一定条件下,用I-还原,产生I2,然后用Na2S2O3标准溶液滴定释放出的I2 。这种方法就叫做间接碘量法或滴定碘法。
间接碘量法也是使用淀粉溶液作指示剂,溶液由蓝色变无色为终点。
注意几点:
1. 酸度的影响:I2 与Na2S2O3应在中性、弱酸性溶液中进行反应。
2. 防止 I2 挥发:?在滴定前,加入过量KI(比理论值大2~3倍),减少 I2挥发:?反应时不可加热;?反应在碘量瓶中进行,滴定时不可过分摇动溶液。
3. 防止I- 被空气氧化 :?避免阳光照射;?滴定快速进行
(二)应用(维生素C含量测定)
维生素C分子中含有还原性的烯二醇基,能被I2定量氧化为二酮基,反应式如下:
C6H8O2 + I2 == C6H6O6 + 2HI
由于反应速率较快,可以直接用I2标准溶液滴定。通过消耗I2溶液的体积及其浓度可以计算试样中维生素C的含量。
由于抗坏血酸具有较强的还原性,在空气极易被氧化而变成黄色,尤其在碱性介质中更甚,测定时加入HAc使溶液呈弱酸性,减少维生素C 副反应,且不影响滴定速度。
由于I2的挥发性及对天平的腐蚀性,不宜在分析天平上称重,故经常先配制一个近似浓度的溶液,然后再进行标定。配制I2溶液时加入过量KI(I2与KI形成KI3使溶解度增加,挥发性大大降低)。溶液保存在棕色瓶中放在暗处,避免见光而使浓度发生改变,还应避免与橡皮等有机物接触。
I2可以用已标定好的Na2S2O3标准溶液来标定I2溶液浓度:
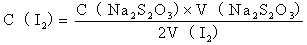
1、配置标准溶液
称取6.5gI2和12.5gKI,置于研钵中,加水研磨溶解后转移至棕色瓶,加水500ml摇匀后避光阴凉处保存。
2、标定I2溶液浓度
吸取25.00mlI2溶液置于250锥形瓶中,加水50ml,用标定过的硫代硫酸钠溶液滴定至浅黄色后,加入淀粉溶液2到4滴,继续滴定至蓝色退去。
3、维生素C含量测定
准确称取约0.2g维生素,放入锥形瓶中,加入新制蒸馏水50ml和10ml 2mol/L HAc溶解,加淀粉指示剂1ml 立即用I2标定至浅蓝色。计算维C含量。
配位滴定法
一、概述
1. 配位滴定法:利用形成配合物的反应进行滴定分析的方法,称为配位滴定法。
2.能够用于配位滴定的反应,必须具备下列条件
(1) 形成的配合物要相当稳定,KMY≥108,否则不易得到明显的滴定终点。
(2) 在一定反应条件下,配位数必须固定(即只形成一种配位数的配合物)。
(3) 反应速度要快。
(4) 要有适当的方法确定滴定的计量点。
目前应用最为广泛的有机配位剂是乙二胺四乙酸(Ethytlene Diamine Tetraacetic Acid简称EDTA)。
二、EDTA配位滴定的基本原理
(一)EDTA配合物的稳定性
水溶液中,EDTA结构式为:
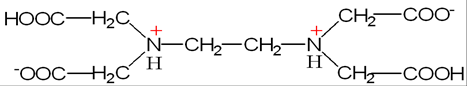
分子中互为对角线的两个羧基上的H+会转移到氮原子上,形成双偶极离子结构。
EDTA与金属离子形成配位化合物的反应如下:
ML (1:1)型配合物
M + L = ML
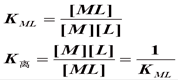
KML越大,配合物越稳定;K离越大,配合物越不稳定。
l EDTA与金属离子形成的配合物具有下列特点
1.配位比较简单,绝大多数为1:1,没有逐级配位的现象。
2.配位能力强,配合物稳定,滴定反应进行的完全程度高。
3.配合物大多带电荷,水溶性较好。
4.配位反应的速率快,除Al、Cr、Ti等金属外,一般都能迅速地完成。
5.配合物的颜色主要决定于金属离子的颜色。即无色的金属离子与EDTA配位,则形成无色的螯合物,有色的金属离子与EDTA配合物时,一般则形成颜色更深的螯合物。如:
NiY2- CuY2- CoY2- MnY2- CrY- FeY-
蓝色 深蓝 紫红 紫红 深紫 黄色
(二)外界条件对EDTA与金属离子配合物稳定性的影响
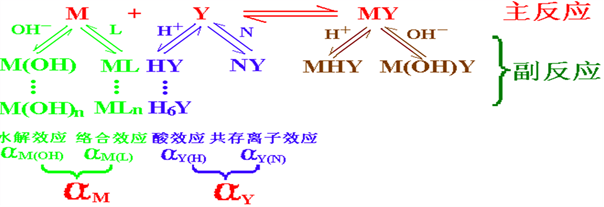
配位滴定中对其影响最大的是酸度以及其他配位剂(络合效应)的影响。
1、酸度的影响
EDTA的酸效应和酸效应系数aY(H)
H+与Y4-离子的副反应对主反应的影响,或由于H+的存在,使配位体Y参加主反应能力降低的现象称为酸效应,也叫质子化效应或pH效应。酸效应的大小,可以用该酸度下,酸效应系数aY(H)来衡量。
滴定时需要满足条件:lgKS?-8≥ lgaY(H)
lgKS?查表可得,它反映EDTA配合物的稳定性,lgKS?越大,配合物越稳定;不同pH下EDTA的lgaY(H)也可以查表得到。
2、其他配位剂的影响(络合效应)
其他络合剂同时存在溶液中时会与金属离子反应,降低金属离子的浓度,配位滴定中常常利用这个原理消除干扰离子。例如,用EDTA滴定Mg2+和Cd2+、Zn2+混合液中的Mg2+时,可以在滴定前先向混合液中加入KCN,使Cd2+、Zn2+与CN-形成稳定的[Zn(CN)4]2-和[Cd(CN)4]2-(Mg2+不与CN-配位),从而消除Cd2+、Zn2+对滴定的干扰。这种消除干扰的作用叫掩蔽作用,起掩蔽作用的配位剂叫掩蔽剂。
(三)金属指示剂
金属指示剂也是一种配位剂,它能与金属离子形成与其本身颜色显著不同的配合物而指示滴定终点。由于它能够指示出溶液中金属离子浓度的变化情况,故也称为金属离子指示剂,简称金属指示剂。
常见的指示剂有铬黑T和钙试剂等,铬黑T(EBT)它是一个三元酸,在溶液中有下列平衡:
H2ln- ←→ HIn2- ←→ In3-
(紫红色) (蓝色) (橙色)
pH<6 pH=7~11 pH>12
铬黑T 在pH<6或pH>12时,游离指示剂的颜色与形成的金属离子配合物的颜色没有显著的差别。只有在pH=7~11时进行滴定,终点由金属离子配合物的红色变成游离指示剂的蓝色,颜色变化才显著。因此,使用金属指示剂,必须注意选用合适的pH范围。
l 金属指示剂必须具备的条件
(1).在滴定的pH范围内,指示剂本身的颜色与其金属离子配合物的颜色应有显著的区别。这样,终点时的颜色变化才明显。
(2).金属离子与指示剂所形成的有色配合物应该足够稳定,在金属离子浓度很小时,仍能呈现明显的颜色,如果它们的稳定性差而离解程度大,则在到达计量点前,就会显示出指示剂本身的颜色,使终点提前出现,颜色变化也不敏锐。
(3)“M—指示剂”配合物的稳定性,应小于“M—EDTA”配合物的稳定性,二者稳定常数应相差在100倍以上,即 logK’MY-logK’MIn>2,这样才能使EDTA滴定到计量点时,将指示剂从“ M—指示剂”配合物中取代出来。
(4).指示剂应具有一定的选择性,即在一定条件下,只对其一种(或某几种)离子发生显色反应。
此外,金属指示剂应比较稳定,便于贮存和使用。
第二篇:电化学实验报告 电位法沉淀滴定测定氯离子的含量.doc已修改
实验三 电位法沉淀滴定测定氯离子的含量
一、目的与要求
掌握电位法沉淀滴定的原理及方法。
二、方法原理
测定水中氯离子的含量,一般用AgNO3溶液滴定,滴定时发生下列反应: Ag+ +Cl- = AgCl↓
在滴定过程中可选用对氯离子或银离子有响应的电极作指示电极。本实验以银电极作指示电极,用带硝酸钾盐桥的饱和甘汞电极作参比电极。银电极的电位与银离子浓度有如下关系:
φAg+/Ag=φ?Ag+/Ag +0.059lgcAg+ (25℃)
随着滴定的进行,银离子浓度逐渐改变,原电池的电动势亦随之变化。根据指示电极电位或电池的电动势对滴定剂体积作图可得到电位滴定曲线,以电位滴定曲线为基础确定滴定终点,根据滴定剂的浓度和所消耗的体积可算出氯离子浓度(或含量)。
三、仪器与试剂
1.数字式酸度计。
2.银电极。
3.饱和甘汞电极。
4.磁力搅拌器。
5.滴定管。
6.CL-离子未知溶液。
7. AgNO溶液:
8.氨水:1+1。
四、内容与步骤
1.硝酸银的标定:
取已知的氯化钠标准溶液15.00ML于100mL 烧杯中,再加约40 mL水。将此烧杯放在磁力搅拌器上,放入搅拌子,然后将清洗后的银电极与玻璃电极,用硝
酸银滴定至终点,计算出硝酸银的浓度。
2. 未知CL-离子含量的测定
1). 用移液管移取15.00 mL CL离子Nacl未知溶液于100mL 烧杯中,再加约-
40 mL水。将此烧杯放在磁力搅拌器上,放入搅拌子,然后将清洗后的银电极玻璃电极,进行测定。
实验操作:
2)打开多功能滴定仪,电脑,点击TitrSation
3) 在多功能滴定仪器上设置
a. 清洗 首先用蒸馏水清洗1~2次,然后用滴定的AgNO3溶液清洗1~2次 b.方法 选择方法,3等当点滴定,确定----编辑方法,模式为0,最大增量0.300 mL,最小增量 0.03 mL,----最大等待时间 5.0秒 最小等待时间 0.0秒----信号漂移值20.00mv/min,极化电压 0mv,预加体积 0ml,电位变化阈值 8.0mv,采集周期2秒-----滴定速度45.0ml/min,等当点 1, 阈值900----安全体积20.00ml ,前三滴加量 0.400ml
c. 样品
d.启动
e. 实验结果保存
f. 数据管理----打开保存数据----存入Excel形式-----查看图形 .
五、数据记录与处理
1. 记录测定水样中氯离子含量时得到的数据,运用φ-V作图法确定终点,计算水样中Cl含量(以mg·L表示)。
六、问题与讨论
(1)试述双盐桥饱和甘汞电极的结构特点及在本实验中的作用?
(2)滴定操作时应注意哪些问题?
--1
-
氧化还原反应实验报告
实验十二氧化还原反应一实验目的1理解电极电势与氧化还原反应的关系和介质浓度对氧化还原反应的影响2加深理解氧化态或还原态物质浓度变化…
-
实验十 氧化还原反应
实验十氧化还原反应一实验目的1加深理解电极电势与氧化还原反应的关系2了解介质的酸碱性对氧化还原反应方向和产物的影响3了解反应物浓度…
-
氧化还原与电化学实验报告
氧化还原与电化学实验报告一实验目的二实验原理三预习思考题1为什么KMnO4能氧化盐酸中的Cl而不能氧化氯化钠溶液中的Cl2为什么H…
- 实验14氧化还原反应
-
氧化还原反应与配位平衡实验报告
氧化还原反应与配位平衡实验报告姓名学号班级实验一氧化还原反应一实验目的学会装配原电池掌握电极的本性电对的氧化型或还原型物质的浓度介…
-
氧化还原反应实验报告
实验十二氧化还原反应一实验目的1理解电极电势与氧化还原反应的关系和介质浓度对氧化还原反应的影响2加深理解氧化态或还原态物质浓度变化…
-
氧化还原与电化学实验报告
氧化还原与电化学实验报告一实验目的二实验原理三预习思考题1为什么KMnO4能氧化盐酸中的Cl而不能氧化氯化钠溶液中的Cl2为什么H…
-
氧化还原反应实验报告
氧化还原反应实验目的通过实验掌握氧化还原反应的基本原理熟悉几种常见的氧化还原反应实验原理物质的氧化还原能力的强弱与物质的本性有关氧…
-
课题_氧化还原反应实验报告
一实验目的1理解电极电势与氧化还原反应的关系和介质浓度对氧化还原反应的影响2加深理解氧化态或还原态物质浓度变化对电极电势的影响3进…
-
南京市雨花台中学氧化还原反应实验报告
实验十二氧化还原反应一实验目的1理解电极电势与氧化还原反应的关系和介质浓度对氧化还原反应的影响2加深理解氧化态或还原态物质浓度变化…
-
分析化学实验报告(武汉大学第五版)
分析化学实验报告陈峻贵州大学矿业学院贵州花溪550025摘要熟悉电子天平的原理和使用规则同时可以学习电子天平的基本操作和常用称量方…