生物化学实验报告:蛋白质分子量的测定——凝胶层析法
实验一 蛋白质分子量的测定——凝胶层析法
一、实验目的
1.掌握凝胶层析的基本原理。
2.学习利用凝胶层析法测定蛋白质相对分子质量的实验技能。
二、实验原理
凝胶层析法也称分子筛层析法,是利用具有一定孔径大小的多孔凝胶作固定相的层析技术。当混合物随流动相经过凝胶层析柱时,其中各组分按其分子大小不同而被分离的技术。该法设备简单、操作方便、重复性好、样品回收率高。
凝胶是一种不带电的具有三维空间的多孔网状结构、呈珠状颗粒的物质,每个颗粒的细微结构及筛孔的直径均匀一致,像筛子,小的分子可以进入凝胶网孔,而大的分子则排阻于颗粒之外。当含有分子大小不一的蛋白质混合物样品加到用此类凝胶颗粒装填而成的层析柱上时,这些物质即随洗脱液的流动而发生移动。大分子物质沿凝胶颗粒间隙随洗脱液移动,流程短,移动速率快,先被洗出层析柱;而小分子物质可通过凝胶网孔进入颗粒内部,然后再扩散出来,故流程长,移动速度慢,最后被洗出层析柱,从而使样品中不同大小的分子彼此获得分离。若分子大小介于上述完全排阻或完全渗入凝胶的物质,则居二者之间从柱中流出。总之,各种不同相对分子质量的蛋白质分子,最终由于它们被排阻和扩散的程度不同,在凝胶柱中所经过的路程和时间也不同,从而彼此可以分离开来。
将凝胶装在柱后,柱床体积称为“总体积”,以Vt表示。实质上Vt是由Vo,Vi与Vg三部分组成,Vo称为“孔隙体积”或“外水体积”,即存在于柱床内凝胶颗粒外面空隙之间的水相体积,相应于一般层析法中柱内流动相的体积;Vi为内体积,即凝胶颗粒内部所含水相的体积。Vg为凝胶本身的体积。洗脱体积(Ve)与Vo与Vi之间的关系可用下式表示:Ve=Vo+KdVi
式中Ve为洗脱体积,自加入样品时算起,到组分最大浓度(峰)出现时所流出的体积;Kd为样品组分在二相间的分配系数,也可以说Kd是分子量不同的溶质在凝胶内部与外部的分配系数。它只与被分离的物质分子的大小和凝胶颗粒孔隙的大小分布有关,而与柱的长度粗细无关,也就是说它对每一物质为常数,与柱的物理条件无关。Kd可通过实验求得,上式可以改写为:Kd=(Ve-Vo)/Vi
上式中Ve为实际测得的洗脱体积;Vo可用不被凝胶滞留的大分子物质的溶液(最好是有颜色以便于观察,如血红蛋白,印度黑墨水,分子量约200万的蓝色葡聚糖-2000等)通过实际测量求得;Vi可由g×Wr求得(g为干胶重量,单位为克;Wr为凝胶的“吸水量”,以毫升每克表示)。因此,对一层析柱胶床来说,只要通过实际实验得知某一物质的洗脱体积就可算出它的Kd值。
如果假定蛋白质分子近于球形,同时没有显著的水合作用,则不同大小分子量的蛋白质,在洗脱时峰的位置和该物质相对分子质量有直接的定量的关系。在一根凝胶柱中,凝胶颗粒间空隙所含水相体积为外水体积Vo,不能进入凝胶孔径的那些大分子,当洗脱体积为Vo时,出现洗脱峰。
凝胶颗粒内部孔穴的总体积为内水体积Vi,能全部渗入凝胶的那些小分子,当洗脱体积为Vo+Vi时,出现洗脱峰。Ve与分子量的关系:对同一类型的化合物,洗脱特性与组分的分子量有关,流过凝胶柱时,按分子量大小顺序流出,分子量大的走在前面。Ve与分子量的关系可用下式表示:Ve=K1-K2logM,式中M为相对分子质量,K1和K2为常数,Ve也可用Ve-Vo。
葡聚糖凝胶有不同的型号,用于分离不同相对分子质量大小的物质。例如Sephardex G-75的分级范围是3000-80000的球形分子。在本实验中用于分离蓝色葡聚糖和牛血清蛋白、胃蛋白酶、CytC。
三、仪器和试剂
1.仪器:层析柱核酸蛋白检测器(或紫外分光光度计)、自动部分收集器、刻度管。
2.试剂:蓝色葡聚糖20##、Sephardex G-75、洗脱液:0.2mol/LTris-HCL-KCL,PH7.5。
3.待测样品:蓝色葡聚糖、牛血清蛋白、胃蛋白酶、CytC。
四、实验步骤
1. 凝胶溶胀
凝胶颗粒要求大小比较均匀 ,可流速稳定,结果较好。取葡聚糖Sephardex G-75干粉,加过量的蒸馏水室温充分溶胀一天(溶胀时间因凝胶交联度不同而不同),或沸水浴中溶胀3小时,这样可大大缩短溶胀时间,而且可以杀死细菌和霉菌,并可排出凝胶内气泡。溶胀过程中注意不要过分搅拌,以防颗粒破碎。待溶胀平衡后用倾泻法除去不易沉下的细小颗粒,最后凝胶经减压抽气除去气泡,即可准备装柱。
2. 装柱与平衡
装柱前须将凝胶上面过多的溶液倾出,将层析柱垂直装好。关闭出口,向柱管内加入约1/3柱容积的洗脱液,然后在搅拌下,将浓浆状的凝胶连续地倾入柱中,使之自然沉降,待凝胶沉降约2—3cm后,打开柱的出口,调节合适的流速,使凝胶继续沉集,待沉集的胶面上升到离柱的顶端约5cm处时停止装柱,关闭出水口。装柱要求连续、均匀、无气泡、无“纹路”。
将洗脱剂与恒流泵相连,恒流泵出口端与层析柱相连。通过2—3倍柱床容积的洗脱液使柱床平衡,然后在凝胶表面上放一片滤纸或尼龙滤布,以防将来在加样时凝胶被冲起,并始终保持凝胶上端有一段液体。
3. 调整洗脱速度
上样前应调整洗脱速度,用自动收集器收集流出液,记录每收集2ml液体所需时间,调整其速度在2min左右。按上述时间设置自动更换收集管。
3. 上样与洗脱
样品上柱是实验成败的关键之一,若样品稀释或上柱不均,会使区带扩散,影响层析效果。上样时应尽量保持床面的稳定。先打开柱的出口,待柱中洗脱液流至距床表面1~2mm时,关闭出口,用滴管将1ml样品慢慢地加至柱床表面,应避免将床面凝胶冲起,打开出口并开始计算流出体积,当样品滲入床中接近床表面1mm时关闭出口。按加样操作,用少量(约1ml)洗脱液冲洗管壁2次。最后加入少量洗脱液于凝胶上,使高出床表面3~5cm,旋紧上口螺丝帽,柱进水口连通恒流泵,开始洗脱。上样的体积,分析用量为柱床容积的1—2%;制备用量为柱床容积的20%~30%。
4. 收集与鉴定
每个收集管内液体用紫外检测仪在280nm波长处检测,出现最高的一个OD值时的体积即为吸收峰的洗脱体积Ve。
5. 凝胶柱的处理
凝胶用过后,反复用蒸馏水洗后保存,如果有颜色或比较脏,可用0.5mol/LNaCl洗涤。短期可保存在水相中,加入防腐剂或加热灭菌后于低温保存。建议长期用干燥状态保存。
五、实验结果

做出洗脱曲线:蓝色葡聚糖-2000
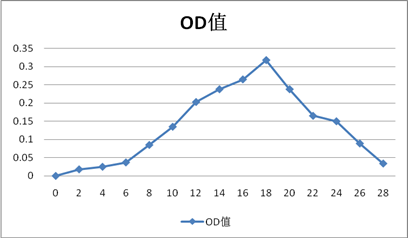
牛血清蛋白
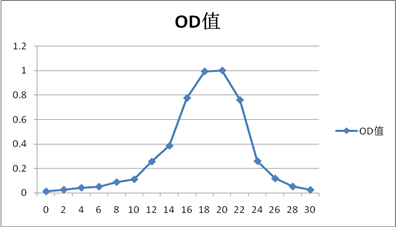
胃蛋白酶

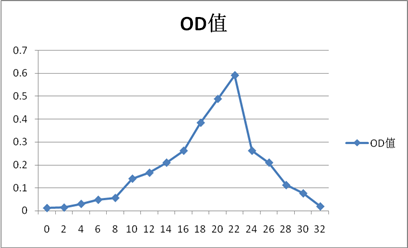
CytC
绘出标准曲线如下: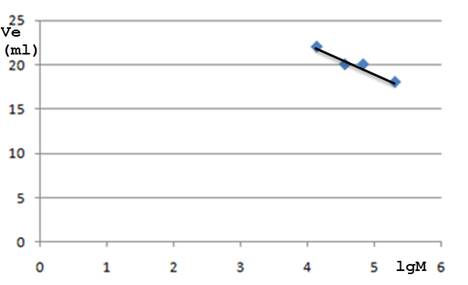
六、注意事项
1.在装柱过程中是需要注意的有以下要求:垂直放置,防止产生气泡,防止柱分层。柱上表面要平,平衡柱时间要长一些,让凝胶充分沉积为均一的柱床。
2.加样前打开出口,使展开剂流出,至正好露出凝胶上平面时,立即关闭出口
3.加样时,用滴管缓缓沿柱内壁旋转加入柱子。
七 、讨论:
1.从洗脱曲线上可以看出,出现了四个较明显的峰值(实验结果较为理想):OD:0.318(Ve=18ml),1.003(Ve=20ml),0.313(Ve=20ml),0.592(Ve=22ml),分别对照试验中所加样的四种蛋白质。由于分子量大的先洗脱出来,小分子量的蛋白后洗脱出来,因此洗脱顺序是蓝色葡聚糖-2000(200KD),牛血清白蛋白(67KD),胃蛋白酶(36KD),Cytc(13.7KD)。可能由于洗脱体积间差距较小,且每管体积为整数,导致牛血清蛋白和胃蛋白酶之间洗脱体积差距未能体现出来。
2.实验过程中,可能由于加样时将凝胶柱的上表面破坏了,导致部分数据不准确,与预期的效果有点误差。
第二篇:蛋白质分子量测定:凝胶过滤层析法
蛋白质分子量测定:凝胶过滤层析法
一、目的:
(1)初步掌握利用凝胶层析法测定蛋白质分子量的原理。
(2)学习用标准蛋白质混合液制作Ve,Kav对1gMr的“选择曲线”以及测定未知蛋白质样品分子量的方法。
二、原理:
凝胶层析法(即凝胶过滤法,gel filtration)是利用凝胶把分子大小不同的物质分离开的一种方法,又叫做分子筛层析法(molecular sieve chromatography),排阻层析法(exclusion chromatography)。凝胶本身是一种分子筛,它可以把分子按大小不同进行分离,好象过筛可以把大颗粒与小颗粒分开一样。但这种“过筛”与普通的过筛不一样。将凝胶颗粒在适宜溶剂中浸泡,使充分吸液膨胀,然后装入层析柱中,加入欲分离的混合物后,再以同一溶剂洗脱,在洗脱过程中,大分子不能进入凝胶内部而沿凝胶颗粒间的空隙最先流出柱外,而小分子可以进入凝胶内部,流速缓慢,以致最后流出柱外,从而使样品中分子大小不同的物质得到分离。分离过程中的示意见图17-1。
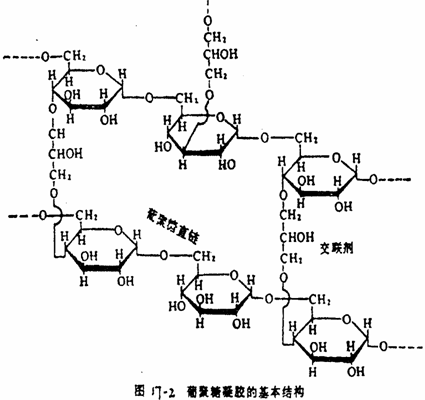
凝胶是由胶体溶液凝结而成的固体物质,不论是天然凝胶还是人工合成凝胶,它们的内部都具有很微细的多孔网状结构。凝胶层析法常用的天然凝胶是琼脂糖凝胶(agarose gel,商品名Sepharose),人工合成凝胶是聚丙烯酰胺凝胶(商品名为Bio-gel-P)和葡聚糖(dextran)凝胶,后者的商品名称为Sephadex型的各种交联葡聚糖凝胶,它是个有不同孔隙度的立体网状结构的凝胶,不溶于水,其化学结构式如图17-2所示。
这种聚合物的立体网状结构,其孔隙大小与被分离物质分子的大小有相应的数量级。在凝胶充分溶胀后,交联度高的,孔隙小,只有相应的小分子可以通过,适于分离小分子物质。相反,交联度低的孔隙大,适于分离大分子物质。利用这种性质可分离不同分子量的物质。
为了说明凝胶层析的原理,将凝胶装柱后,柱床体积称为“总体积”,以Vt(total volume)表示。实际上Vt是由Vo,Vi与Vg三部分组成,即:
Vt=Vo+Vi+Vg
Vo称为“孔隙体积”或“外体积”(outer volume)又称“外水体积”,即存在于柱床内凝胶颗粒外面空隙之间的水相体积,相应于一般层析法中柱内流动相的体积;Vi为内体
积(inner volume),又称“内水体积”,即凝胶颗粒内部所含水相的体积,相应于一般层析法中的固定相的体积,它可从干凝胶颗粒重量和吸水后的重量求得;Vg为凝胶本身的体积,因此Vt—Vo等于Vi+Vg 。它们之间的关系可用图17-3表示。洗脱体积(Ve,elution Volume)与Vo及Vi之间的关系可用下式表示:
Ve=Vo+KdVi
式中Ve为洗脱体积,自加入样品时算起,到组分最大浓度(峰)出现时所流出的体积;Kd为样品组分在二相间的分配系数,也可以说Kd是分子量不同的溶质在凝胶内部和外部的分配系数。
它只与被分离物质分子的大小和凝胶颗粒孔隙的大小分布有关,而与柱的长短粗细无关,也就是说它对每一物质为常数,与柱的物理条件无关。Kd可通过实验求得,上式可改写成:
上式中Ve为实际测得的洗脱体积;Vo可用不被凝胶滞留的大分子物质的溶液(最好有颜色以便于观察,如血红蛋白,印度黑墨水,分子量约200万的蓝色葡聚糖-2000等)通过实际测量求出;Vi可由g·WR求得(g为干凝胶重,单位为克;WR为凝胶的“吸水量”,以毫升/克表示)。因此,对一层析柱凝胶床来说,只要通过实验得知某一物质的洗脱体积Ve,就可算出它的Kd值。以上关系可用图17-4表示。
Kd可以有下列几种情况:
有颜色以便于观察,如血红蛋白,印度黑墨水,分子量约200万的蓝色葡聚糖-2000等)通过实际测量求出;Vi可由g·WR求得(g为干凝胶重,单位为克;WR为凝胶的“吸水量”,以毫升/克表示)。因此,对一层析柱凝胶床来说,只要通过实验得知某一物质的洗脱体积Ve,就可算出它的Kd值。以上关系可用图17-4表示。
Kd可以有下列几种情况:
1、当Kd=0时,则Ve=Vo。即对于根本不能进入凝胶内部的大分子物质(全排阻),洗脱体积等于空隙体积(图中组分Ⅰ)。
2、当Kd=1时,Ve=Vo+Vi。即小分子可完全渗入凝胶内部时,洗脱体积应为空隙体积与内体积之和(图中组分Ⅲ)。
可以看出,对某一凝胶介质,两种全排出的分子即Kd都等于零,虽然分子大小有差别,但不能有分离效果。同样两种分子如都能进入内部空隙,即Kd都等于1,它们既使分子大小有不同,也没有分离效果。因此不同型号的凝胶介质,有它一定的使用范围。
3、当0<Kd<1时,Ve=Vo+KdVi。表示内体积只有一部分可被组分利用,扩散渗入,Ve即在Vo+Vi之间变化(图中组分Ⅱ)。
4、有时Kd>1,表示凝胶对组分有吸附作用,此时Ve>Vo+Vi。例如一些芳香族化合物的洗脱体积远超出理论计算的最大值,这些化合物的Kd>1。如苯丙氨酸,酪氨酸和色氨酸在Sephadex G-25中的Kd值分别为1.2,1.4和2.2。
在实际工作中,对小分子物质也得不到Kd=1的数值,特别是交联度大的凝胶差别更大,如用G-10型得Kd值0.75左右,用G-25型得0.8左右。这是由于一部分水相与凝胶结合较牢固,成为凝胶本身的一部分,因而不起作用,小分子不能扩散入内所致。此时Vi即不能以g·WR计算,为此也常有直接用小分子物质D2O,NaCl等通过凝胶柱而由实验计算出Vi值的。另一个解决的办法是不使用Vi与Kd,而用Kav(有效分配系数)代替Kd,其定义如下:
已知
在这里实际上将原来以水作为固定相(Vi)改为水与凝胶颗粒(Vi—Vo)作为固定相,而洗脱剂(Ve—Vo)作为流动相。Kav与Kd对交联度小的凝胶差别较小,而对交联度大的凝胶差别大。
在一般情况下,凝胶对组分没有吸附作用时,当流动相流过Vt体积后,所有的组分都应该被洗出来,这一点为凝胶层析法的特点,与一般层析方法不同。
Ve与分子量的关系:对同一类型的化合物,洗脱特性与组分的分子量有关,流过凝胶柱时,按分子量大小顺序流出,分子量大的走在前面。Ve与分子量的关系可用下式表示:
V e=K1—K2logMr
K1与K2为常数,Mr为分子量,Ve也可用Ve—Vo(分离体积),Ve/Vo(相对保留体积),Ve/Vt(简化的洗脱体积,它受柱的填充情况的影响较小)或Kav代替,与分子量的关系同上式,只是常数不同。通常多以Kav对分子量的对数作图得一曲线,称为“选择曲线”,如图17-5所示。曲线的斜率是说明凝胶性质的一个很重要的特征。在允许的工作范围内,曲线愈陡,则分级愈好,而工作范围愈窄。凝胶层析主要决定于溶质分子的大小,每一类型的化合物如球蛋白类,右旋糖酐类等都有它自己的特殊的选择曲线,可用以测定未知物的分子量,测定时以使用曲线的直线部分为宜。
用凝胶层析法测定蛋白质的分子量,方法简单,技术易掌握,样品用量少,而且有时不需要纯物质,用一粗制品即可。例如在一粗酶制剂中,为了测定某一酶组分的分子量,只要测定洗脱液中具有该酶最大活性的部分,然后确定其洗脱体积,即可从标准曲线中查出其分子量。
凝胶层析法测定分子量也有一定的局限性,在pH6—8的范围内,线性关系比较好,但在极端pH时,一般蛋白质有可能因变性而偏离。糖蛋白在含糖量超过5%时,测得分子量比真实的要大,铁蛋白则与此相反,测得的分子量比真实的要小。
有一些酶底物是糖,如淀粉酶,溶菌酶等会与葡聚糖凝胶形成络合物,这种络合物与酶-底物络合物相似,因此在葡聚糖凝胶上层析时表现异常。用凝胶层析法所测分子量的结果,要和其它方法的测定相对照,由此才可作出较可靠的结论。
凝胶层析技术操作方便,设备简单,周期短,重复性能好,而且条件温和,一般不引起生物活性物质的变化,目前已得到相当广泛的应用,例如可用于脱盐,浓缩高分子物质的溶液,生化物质的分离提纯,去除热源物质以及用于测定高分子物质的分子量。下面实验是用葡聚糖凝胶层析法测定蛋白质的分子量。
三、试剂及器材:
1、试剂:
(1)标准蛋白质混合液(各2—3mg·ml-1,用氯化钾-乙酸溶液配制),牛血清清蛋白(Mr67000),鸡卵清蛋白(Mr43000),胰凝乳蛋白酶原A(Mr25000)和结晶牛胰岛素(PH2时为二聚体Mr12000)等均需层析纯。
(2)蓝色葡聚糖-2000(8—10mg·ml-1),N-乙酰酪氨酸乙酯(或硫酸铵或重铬酸钾)(8—10mg·ml-1)。
(3)0.025mo·L-1氯化钾-0.2mol·L-1乙酸溶液,Sephadex G—75(或G—100),5%Ba(Ac)2。
(4)待测蛋白质样品液。
2.器材:
层析柱:柱管(直径1.0~1.3cm;管长90~100cm),核酸蛋白检测仪或紫外分光光度计,自动部分收集器,恒压瓶,下口瓶。
四、操作步骤:
1、一般操作方法:
(1)凝胶的选择和处理
凝胶颗粒最好大小比较均匀,这样,流速稳定,结果较好。如果颗粒大小不匀,用倾泻法倾去不易沉下的较细颗粒。
将称好的干粉倾入过量的洗脱液(一般多用水,盐溶液或缓冲溶液)中,室温下放置,使之充分溶胀。注意不要过分搅抖,以防颗粒破碎。溶胀时间因凝胶交联度不同而异为了缩短溶胀时间,可在沸水浴上加热将近100℃,这样可大大缩短溶胀时间至几小时,而且还可以杀死细菌和霉菌,并可排除凝胶内气泡。种类凝胶溶胀时间请查阅葡聚糖凝胶的技术数据表。
(2)装柱
装柱前,必须用真空干燥器抽尽凝胶中空气。装柱方法与一般柱层析法相似。层析柱可以自制或外购,目前已有各种规格层析柱商品(图17-6)。
装柱前须将凝胶上面过多的溶液倾出。关闭层析柱出水口,并向柱管内加入约1/3柱容积的洗脱液,然后在搅拌下,将浓浆状的凝胶连续地倾入柱中,使之自然沉降,待凝胶沉降约2—3cm后,打开柱的出口,调节合适的流速,使凝胶继续沉集,待沉集的胶面上升到离柱的顶端约5cm处时停止装柱,关闭出水口。接着再通过2—3倍柱床容积的洗脱液使柱床稳定,然后在凝胶表面上放一片滤纸或尼龙滤布,以防将来在加样时凝胶被冲起,并始终保持凝胶上端有一段液体。
新装好的柱要检验其均一性,可用带色的高分子物质如蓝色葡聚糖-2000(又称蓝色右旋糖,商品名为Blue dextran-2000)、红色葡聚糖或细胞色素C等配成2mg/ml的溶液过柱,看色带是否均匀下降,或将柱管向光照方向用眼睛观察,看是否均匀,有无“纹路”或气泡。若层析柱床不均一,必须重新装柱。
(3)加样
要照顾到浓度与粘度两个方面。分析用量:1—2ml/100ml柱床容积(1—2%);制备用量:20—30ml/100ml柱床容积。
加样方法与一般柱层析相同。夹紧上、下进、出水口夹子,为防止操作压改变,可将塑料管下口抬高至离柱上端约50cm处(Sephadex G-75,选用50cm液柱操作压),打开柱上端的塞子或螺丝帽,吸出层析柱中多余液体直至与胶面相切。沿管壁将样品溶液小心加到凝胶床面上,应避免将床面凝胶冲起,打开下口夹子,使样品溶液流入柱内,同时收集流出液,当样品溶液流至与胶面相切时,夹紧下口夹子。按加样操作,用1ml洗脱液冲洗管壁2次。最后加入3—4ml洗脱液于凝胶上,旋紧上口螺丝帽,柱进水口连通恒压瓶,柱出水口与核酸蛋白质检测仪比色池进液口相连,比色池出液口再与自动部分收集器相连(如图17-7)。
(4)洗脱
洗脱液应与凝胶溶胀所用液体相同。洗脱用的液体有水(多用于分离不带电荷的中性物质)及电解质溶液(用于分离带电基团的样品),如酸、碱、盐的溶液及缓冲液等。对于吸附较强的组分,也有使用水与有机溶剂的混合液,如水-甲醇、水—乙醇、水-丙酮等为洗脱剂,可以减少吸附,将组分洗下。本实验洗脱用0.025mol·L-1氯化钾-0.2mol·L-1乙酸溶液。
洗脱时,打开上、下进出口夹子,用0.025mol·L-1氯化钾-0.2mol·L-1乙酸溶液,以每管3ml/10min流速洗脱,用自动部分收集器收集流出液。
影响洗脱液流速的因素有:
①洗脱液加在柱上的压力——操作压(由液面差引起)。一般来说,流速与柱压力差成正比,但对某些种类凝胶加压不能超过一个极限值,否则加大压力,不仅不能增加流速,反而使流速急剧下降,特别是在使用交联度小(WR>7.5)的凝胶时要特别注意。为保持层析过程中恒定的压力,可使用恒压瓶。
②凝胶交联度;交联度大的凝胶(G-10至G-50型)的流速与柱的压力差成正比,与柱长成反比,与柱的直径无关。交联度小的凝胶(G-75 至G-200型)的流速与柱的直径有关,并在一定操作压差下有最大的流速值,操作压见图17-8。
③凝胶颗粒大小:颗粒大时,流速较大,但流速大时洗脱峰形常较宽。颗粒小时,流速较慢,分离情况较好。要求在不影响分离效果情况下,尽可能使流速不致太慢,以免用时过长。
(5)重装
一般地说,一次装柱后,可反复使用,无特殊的“再生”处理,只须在每次层析后用3—4倍柱床体积的洗脱液过柱。由于使用过程中,颗粒可能逐步沉积压紧,流速逐渐会减低,使得一次分析用时过多,这时需要将凝胶倒出,重新填装;或用反冲方法,使凝胶松动冲起,再行沉降。有时流速改变是由于凝胶顶部有杂质集聚,则需竟混有脏物的凝胶取出,必要时可将上部凝胶搅松后补充部分新胶,经沉集、平衡后即可使用。
(6)凝胶的保存方法
凝胶用完后,可用以下方法保存。
①膨胀状态:即在水相中保存。可按注意事项(9),加入防腐剂或加热灭菌后于低温保存。
②半收缩状态:用完后用水洗净,然后再用60%—70%乙醇洗,则凝胶体积缩小,于低温保存。
③干燥状态:用水洗净后,加入含乙醇的水洗,并逐渐加大含醇量,最后用95%乙醇洗,则凝胶脱水收缩,再用乙醚洗去乙醇,抽滤至干,于60—80℃干燥后保存。这3种方法中,以干燥状态保存为最好。
2.蛋白质分子量测定
根据待测蛋白质的分子量范围选用Sephadex G-75或G-100型凝胶,其颗粒在40—120μm,柱管选用直径1.0—1.3cm,柱长90—100cm,本实验选用1.1×100cm商品层析柱。
(1)测定Vo和Vi
将0.5ml蓝色葡聚糖-2000和硫酸铵混合液(2mg·ml-1)上柱、洗脱,分别测出Ve。蓝色葡聚糖的Ve即为该柱的Vo,硫酸铵洗脱体积Ve减去Vo即为柱的Vi。蓝色葡聚糖的洗脱峰可根据颜色判断,硫酸铵洗脱峰用Ba(Ac)2生成沉淀判断(若用重铬酸钾,其洗脱峰,可根据颜色判断)。
(2)标准曲线的制作
按上述方法将1ml标准蛋白质混合液上柱,然后用0.025mol·L-1氯化钾-0.2mol·L-1乙酸溶液洗脱。流速3ml/10min,3ml/管。用部分收集器收集,核酸蛋白质检测仪280nm处检测,记录洗脱曲线,或收集后用紫外分光光度计280nm处测定每管光吸收值。以管号(或洗脱体积)为横坐标,光吸收值为纵坐标作出洗脱曲线。
根据洗脱峰位置,量出每种蛋白质的洗脱体积(Ve)。然后,以蛋白质分子量的对数1gMr为纵坐标,Ve为横坐标,作出标准曲线(图17-9)。为了结果可靠,应以同样条件重复1—2次,取Ve的平均值作图。同时根据已测出的Vo和Vi以及通过测量柱的直径和凝胶柱床高度,计算出的Vt,分别求出Kd和Kav。
也可以Kd或Kav为横坐标,1gMr为纵坐标作出标准曲线。
(3)未知样品分子量的测定
完全按照标准曲线的条件操作。
五、结果与讨论:
根据紫外检测的洗脱峰位置,量出洗脱体积,重复测定1—2次,取其平均值,也可以计算出Kav,分别由标准曲线查得样品的分子量。
注 意 事 项
(1)根据层析柱的容积和所选用的凝胶溶胀后柱床容积,计算所需凝胶干粉的重量,以将用作洗脱剂的溶液使其充分溶胀。
(2)层析柱粗细必须均匀,柱管大小可根据实际需要选择。一般来说,细长的柱分离效果较好。若样品量多,最好选用内径较粗的柱,但此时分离效果稍差。柱管内径太小时,会发生“管壁效应”,即柱管中心部分的组分移动慢,而管壁周围的移动快。柱越长,分离效果越好,但柱过长,实验时间长,样品稀释度大,分离效果反而不好。
用于脱盐的柱一般都是短而粗,柱长(L)/直径(D)<10;对分级分离用的柱,L/D值可以比较大,对很难分离的组分可以达到L/D=100,一般选用内径为1cm,柱长100cm就够了。
(3)各接头不能漏气,连续用的小乳胶管不要有破损,否则造成漏气、漏液。注意恒压瓶内的排气管应无液体,并随着柱下口溶液的流出不断有气泡产生,则表示恒压瓶不漏气。操作过程中,层析柱内液面不断下降,则表示整个系统有漏气之处,应仔细检查并加以纠正。
(4)装柱要均匀,不过松也不过紧,最好也在要求的操作压下装柱,流速不宜过快,避免因此而压紧凝胶。但也不宜过慢,使柱装得太松,导致层析过程中,凝胶床高度下降。
(5)始终保持柱内液面高于凝胶表面,否则水分挥发,凝胶变干。也要防止液体流干,使凝胶混入大量气泡,影响液体在柱内的流动,导致分离效果变坏,不得不重新装柱。
(6)样品溶液的浓度和粘度要合适。浓度大,自然粘度增加。一个粘度很大的样品上柱后,样品分子因运动受限制,影响进出凝胶孔隙,洗脱峰形显得宽而矮,有些可分离的组分也因此重叠。
(7)洗脱用的液体应与凝胶溶胀所用液体相同,否则,由于更换溶剂引起凝胶容积变化,从而影响分离效果。
(8)操作压过大可使流速变慢,此外还可以导致凝胶胶面下降,柱床容积的改变,相应测出Vt,Vo,Ve等也改变,造成实验结果的误差。
(9)由于葡聚糖凝胶为糖类化合物,并且一定要在液相中操作,所以有必要注意防止发霉与生长细菌。一般可用0.02%的叠氮钠(NaN3)溶液,它极易溶于水,不与蛋白质和糖反应,也不改变它们的层析效果,也可用0.002%的洗必泰(hibitane)溶液。在弱酸性溶液中,可用0.05%(或0.01%—0.02%)三氯丁醇溶液,但它在强碱性溶液或加热到60℃以上时就要分解。在弱碱性溶液中也可用0.01%乙酸汞溶液防腐消毒。一般不用氯仿、丁醇、甲苯等为防腐剂。因为它们能引起凝胶颗粒的收缩,同时还能进入层析仪器的塑料部件,使塑料变软,造成不良后果。
(10)使用部分收集器时,将其转盘调节到最外圈,从第一管开始,否则会造成转换臂的转动与转盘不同心,以致洗脱液流到管外。
凝胶层析中常遇到的故障之原因与排除方法总结见表17-1。
故 障
产 生 的 原 因
排 出 的 方 法
恒压瓶不能恒压
1.恒压瓶上口或下口橡胶塞未塞紧或橡胶塞插玻璃管处漏气
1.找出漏气原因,塞紧橡胶塞
层析柱连接后,进水口无液体滴出
2.层析柱进水口或出水口的止水夹未打开
3.进水口塑料管中有气泡
2.打开止水夹
3.排除塑料管中的气泡
层析柱出水口无液体流出
4.出水口的止水夹未打开
5.层析柱下口螺丝未旋紧,因漏气而造成出水塑料管中有气泡
6.出水口塑料管被凝胶阻塞
4.打开出水口止水夹并调节流速
5.找出产生气泡的原因,排除气泡
6.将层析柱中的凝胶倒出,冲洗尼龙网排除塑料管中的凝胶,重新装柱
层析过程中流速逐渐减慢
7.样品中或洗脱缓冲液中含有不溶颗粒将胶床表面阻塞
8.操作压过高,将凝胶胶床压紧
9.测定内水时硫酸铵浓度太大;凝胶脱水使胶床压紧
10.加样时未注意恒压,加样后胶床床面下降
11.长期使用,微生物生长
12.装柱时凝胶未完全溶胀,平衡时流速即逐渐减慢
13.凝胶颗粒过细,或由于用暴力搅动凝胶使凝胶颗粒打碎
7.采用离心或过滤法除去不溶颗粒,用滴管移去柱床表面1—2cm的凝胶,补加新凝胶至同样高度
8.重新装柱,采用适当的操作压
9.将凝胶取出用缓冲液反复洗涤,溶胀重新装柱。适当降低硫酸铵浓度
10.加样时应根据操作压,将塑料管下水口抬高至相应的操作压
11.层析柱不用时,在平衡缓冲液中加入0.02%叠氮钠或0.002%洗必泰,并使其充满柱床体积,以抑制细菌生长,暂时不用的柱应定期用缓冲液过柱冲洗,也可以防止微生物生长
12.将凝胶取出,待其完全溶胀后重新装柱
13.取出层析柱中的凝胶,用漂浮法除去过细的颗粒,重新装柱,搅拌凝胶应防止暴力
层析柱胶床中有气泡
14.装柱前,凝胶未抽气或煮沸,在凝胶中混入空气
15.加样不当使空气进入凝胶胶床
16.从冰箱中取出凝胶或凝胶缓冲液立即装柱,或装柱后被太阳暴晒
14.在凝胶柱上层的气泡可用细头长滴管或细塑料管将气泡取出或赶走,并重新平衡,稳定胶床后再使用,若气泡太多则应抽气或煮沸后自然冷却后装柱
15.小心加样防止带进气泡
16.凝胶或缓冲液应放置到室温后才能装柱,避免太阳直射
层析柱胶床破裂
17.大量空气进入层析柱
18.进水口流速慢,出水口流速快
17.找出空气进入层析柱的原因,重新装柱
18.找出进出水口流速不一致的原因,重新装柱
故 障
产 生 的 原 因
排 队 的 方 法
样品进入凝胶后条带扭曲
19.样品或缓冲液中有颗粒或不溶物
20.凝胶表面不平,或胶床不均匀
19.按方法7处理样品或缓冲液
20.将凝胶柱置于垂直位,轻轻搅动胶床表面1—2cm处的凝胶,使其自然沉降
凝胶层析分辨率不高
21.凝胶层析柱装得不均匀
22.凝胶G型选择不当
23.加样量太大
24.柱床太短
25.样品浓度高,粘度大而形成拖尾
26.洗脱时流速太快
27.分部收集时每管体积过大
21.将凝胶取出重新装柱
22.根据欲分离物质分离的情况,选择合适的凝胶G型与粒度
23.为提高分辨率,分析时加样量一般为柱长的1—2%,最多不能超过5%
24.将柱床高度适当加长
25.根据紫外测定的光吸收值将样品适当稀释
26.调节洗脱的流速,(本实验为3ml/10min)
27.控制每管收集量,为便于紫外测定,每管收集量以2.8—3ml为宜
分部收集时,收集盘转动一次间隔数只试管或滴管口偏离相应的试管
28.使用前未经定位或开始收集后随便移动转换臂,从而造成收集盘与转换臂转动不同步
28.将自动收集器换向开关拨向“顺”,连续按手动开关,使收集盘与转换臂同时以“顺”时针方向转至外圈零位。将换向开关拨至“逆”,使转换臂的滴管口对准第一个试管中心,打开自动开关即可进行同步收集
-
蛋白质的定量测定实验报告
显问题其次回顾全部过程的原理我们猜测可能存在的造成结果低的情况为在室温冷却后实验室没有空余的分光光度计排队时间应该是全部小组中最长…
-
蛋白质测定实验报告
蛋白质测定方法化学报告蛋白质的检测酚试剂法灵敏度较高20250mg费时蛋白质在碱性溶酚类柠檬液中其肽键与酸硫酸铵Cu2螯合形成tr…
-
蛋白质测定实验报告
生物化学实验报告姓名学号专业年级组别生物化学与分子生物学实验教学中心实验名称实验日期20xx1015合作者评分Folin酚试剂法测…
-
紫外分光光度法测定蛋白质含量实验报告
紫外分光光度法测定蛋白质含量一实验目的1学习紫外光度法测定蛋白质含量的原理2掌握紫外分光光度法测蛋白质含量的实验技术二实验原理1测…
-
分光法测蛋白质实验报告
紫外分光光度法测定蛋白质含量一实验目的1学习紫外分光光度法测定蛋白质含量的原理2掌握紫外分光光度法测定蛋白质含量的实验技术3掌握T…
-
蛋白质测定实验报告
蛋白质测定方法化学报告蛋白质的检测酚试剂法灵敏度较高20250mg费时蛋白质在碱性溶酚类柠檬液中其肽键与酸硫酸铵Cu2螯合形成tr…
-
蛋白质功能性质的检测实验报告
华南农业大学实验报告专业班次组别题目蛋白质功能性质的检测姓名黄俊怡日期20xx0711一实验目的通过本实验定性地了解蛋白质的主要功…
-
SCAU实验报告蛋白质功能性质的检测
蛋白质功能性质的检测一实验目的通过本实验定性地了解蛋白质的主要功能性质二实验原理蛋白质的功能性质一般是指能使蛋白质成为人们所需要的…
-
实验报告 蛋白质分子的测定
实验一蛋白质分子的测定凝胶层析法一实验原理凝胶层析法即凝胶过滤法gelfiltration是利用凝胶把分子大小不同的物质分离开的一…
-
蛋白质的定量测定实验报告
显问题其次回顾全部过程的原理我们猜测可能存在的造成结果低的情况为在室温冷却后实验室没有空余的分光光度计排队时间应该是全部小组中最长…
-
生化实验报告 福林-酚试剂法测定蛋白质的浓度
实验三福林Folin酚试剂法测定蛋白质的浓度一实验目的1学会制备无蛋白血滤液2掌握FolinWu法测定血糖含量的原理和方法3掌握7…