实验一 蛋白质含量测定方法的研究
生物化学实验预习报告
实验一 蛋白质含量测定方法的研究
一、研究背景
蛋白质含量的测定广泛应用于生产实践、医药卫生等各个领域,如测定牛奶等食品中蛋白质的含量作为其营养成分的参考值,测定病人尿液中蛋白质的含量以检测其是否患有某种疾病等,因而测定蛋白质的含量具有非常重要的意义。由于蛋白质本身具有一系列的特点(如具有等电点、富含有机氮、含有肽键、存在某些含共轭双键的氨基酸残基、可与某些染料结合等),利用它的这些特点可以通过特定的化学反应或者某些物理手段,将一定量的蛋白质转化为可以方便定量测定的其他量,从而达到测定蛋白质含量的目的。蛋白质诸多特点的存在决定了它会有不同的测定方法,同时由于多种非蛋白组分的存在和干扰以及各种方法本身的局限性(适用条件),每一种测定方法都有各自的优缺点,某一种方法并不能在任何条件下适用于任何形式的蛋白质,所以多种测定方法的共存是有意义的。目前常用的蛋白质含量测定方法有四种:凯氏定氮法、Folin-酚法、考马斯亮蓝(G-250)法、紫外法,其中后三种方法最常用。在实际工作中,需要根据所测定对象的具体情况综合考虑各种因素(如实验器材的限制、实验所要求的灵敏度和精确度、所测定蛋白质的种类与性质、溶液中存在的干扰物质、测定所花费的时间等),有选择性地使用某种方法。
二、研究目标
1.掌握常用的蛋白质含量测定方法的原理和操作流程;(基础目标)
2.了解不同测定方法的优点和局限性,掌握在实际工作中不同测定方法的选择条件;(基础目标)
3.证明非蛋白组分在蛋白质含量测定中存在干扰,并通过分析实验结果确定该干扰在实际工作中是否可以忽略。(高级目标)
三、研究策略
本实验最重要的目的是为了确定非蛋白组分在蛋白质含量测定中存在的干扰是否可以忽略。思路有两种:一种方法是先测定纯蛋白质溶液中蛋白质含量,随后在该溶液中加入非蛋白组分(如嘌呤、嘧啶等吸光物质,Ca2+等金属离子,无机酸或无机碱,甚至某些色素等),测定此时蛋白质含量,对比两次测定结果,计算相对误差,从而得出非蛋白组分的影响程度;另一种方法是直接使用天然的蛋白质粗提取物(含有非蛋白组分,并将其中包含的所有非蛋白组分看成一个影响因素),来测定粗提取物中的蛋白质含量。考虑到第一种方法中涉及到的非蛋白组分的加入量没有标准,这些非蛋白材料(影响因素)实验室内也不一定有,更重要的是该实验所具有的实际意义不大,故决定采用第二种方案。但是方案二所存在的一个问题是所测组分蛋白质含量的真实值未知,测得的结果没有可以参考比较的依据,所以需要另外找一个可以参考的标准。
所采取的策略是先用不同的方法测定已知标准蛋白质溶液的浓度,三个测定值之间会有一定的相关性(或者说测定值之间的变化符合某种趋势),随后测定天然样品蛋白质粗提取液中蛋白含量,得到另外三个测定值,这三个测定值之间同样会存在一定的相关性。如果这两个相关性关系(或者说变化趋势)大致相同,则认为非蛋白组分的影响很小,可以忽略,相反则不能忽略。具体分以下五步走:
1.分别用紫外法、Folin-酚法、考马斯亮蓝(G-250)法对标准牛血清白蛋白溶液绘制标准曲线;
2.结合各标准曲线,用三种方法分别测定待测样液1(待测液1A,500μg/ml BSA、待测液1B,250μg/ml BSA、待测液1C,100μg/ml BSA、待测液1D,10μg/ml BSA)各组中蛋白质的含量;
3.用三种方法分别测定待测样液2(生物样品的蛋白质粗提取液,含非蛋白组分)的蛋白质含量;
4.对比分析在不同稀释倍数下使用各种方法测定的样液1中各组蛋白质的含量值,与真实值相比较并计算相对误差,得出不同测定方法的精确度或者所适宜测定的蛋白质浓度范围;
5.将不同方法测定的样液1及样液2蛋白质含量值分别绘制成曲线图,对比分析两条曲线的波动趋势或相似程度,若波动趋势大致相同,则说明非蛋白组分在蛋白质含量测定中的影响可以忽略;若波动趋势明显相差较大,则非蛋白组分在蛋白质含量测定中的影响不能忽略。
四、研究方案及可行性分析
1.实验原理与技术
(1)凯氏定氮法
原理:样品中含氮有机化合物与浓硫酸在催化剂作用下共热消化,含氮有机物分解产生氨,氨又与硫酸作用,变成硫酸铵。然后加碱蒸馏放出氨,氨用过量的硼酸溶液吸收,再用盐酸标准溶液滴定求出总氮量换算为蛋白质含量。总体上分为样品消化、蒸馏、吸收和滴定四个过程。
优点:是一种蛋白质的经典测定方法,适用广泛,可用于动植物各种组织、器官及食品等组成复杂的样品测定;测定结果准确,重现性好,往往将该方法测定的蛋白质作为其他方法的标准蛋白,该方法也是目前分析有机化合物含氮量最常用的方法。
缺点:灵敏度低,0.2~1.0mg/ml;蒸馏与吸收操作装置较复杂,不便于操作;实验过程所需时间较长,操作费时;因实验过程会产生有毒气体,通常需在通风橱中进行样品消化,另外样品与浓H2 SO4 共热,有一定危险。
基于上述操作的不便性及危险性,本次实验不使用该方法测定蛋白质含量。
(2)紫外法
原理:在蛋白质分子中,苯丙氨酸、酪氨酸和色氨酸残基的苯环含有共轭双键,使蛋白质具有吸收紫外光的性质,且吸收峰在280nm处,在该波长范围内,其吸光度(即光密度值OD280)与蛋白质含量(浓度)呈正比关系,故可进行蛋白质含量的测定。
优点:简便、灵敏、快速,50~100μg/ml;低浓度盐类不干扰测定;不消耗样品,测定后样品仍能回收使用。
缺点:准确度较差,干扰物质多。在用标准曲线法测定蛋白质含量时,对那些与标准蛋白质中酪氨酸、色氨酸含量差异较大的蛋白质有一定的误差,故该方法适用于测定与标准蛋白氨基酸组成相似的蛋白质;若样品中含有嘌呤、嘧啶以及核酸等吸收紫外光的物质时,会出现较大的干扰,需进行矫正,但不同蛋白质及核酸的紫外吸收值不同,虽经过矫正但还是存在一定的误差;在测定时,由于蛋白质吸收高峰常因pH的改变而有变化,因此需要注意溶液的pH,且测定样品时pH要与测定标准曲线时相一致;该方法适于测无色样品,对有色样品需进行预处理,排出颜色干扰之后才能使用此方法。[1]
(3)Folin-酚法(Lowry法)
原理:该方法根据蛋白质侧链基团中的特殊残基进行含量测定,其基础是蛋白质中所含的酪氨酸、色氨酸等残基数目与蛋白质含量成正比。首先由Folin-酚试剂甲(相当于双缩脲试剂)中的Cu++离子在碱性条件(pH10)下与蛋白质中的肽键形成络合物,然后加入试剂乙(磷钨酸和磷钼酸的混合液)使其被蛋白质中的含有酚基的氨基酸残基还原为钼蓝和钨蓝的混合物,呈现出蓝色反应。反应颜色的深浅与蛋白质含量呈正比,据此可测定蛋白质的含量。
优点:反应灵敏,10~200μg/ml,灵敏度为紫外法的10~20倍,为双缩脲反应的100倍;操作简便,适于小规模分析。
缺点:稍微有些费时,且需要严格控制时间;显色量随蛋白质种类的不同而有差异;与蛋白质的反应为非特异性反应,抗干扰能力较差,如Mg2+、Tris缓冲液、甘油、EDTA等均会产生干扰。
(4)考马斯亮蓝(G-250)法(Bradfold法)
原理:根据蛋白质与染料相结合的原理而设计。在酸性溶液中,考马斯亮蓝G-250染料与蛋白质相结合,使染料的最大吸收峰的位置由465nm变为595nm,溶液的颜色也由棕黑色变为蓝色。有研究认为,染料主要是与蛋白质中的碱性氨基酸(特别是精氨酸)和芳香族氨基酸残基相结合。在波长为595nm下的吸光度值A595与蛋白质浓度呈正比,故可用于蛋白质的定量测量。
优点:是目前灵敏度最高的蛋白质含量测定方法,可精确测量到1μg/ml;测定快速简便,只需要一种试剂,蛋白质与染料结合2min左右就可达到平衡,且结合物在1h内保持稳定;干扰物质少,干扰Lowry法的K+ 、Na + 、Mg2+离子、Tris缓冲液、蔗糖、甘油、巯基乙醇、EDTA等均不干扰该测定方法。故该方法适用于灵敏度要求高、快速定量测定微量蛋白质的测定。[2]
缺点:由于各种蛋白质中的精氨酸和芳香族氨基酸的含量不同,因此Bradfold法用于不同蛋白质测定时会有较大的偏差,在制作标准曲线时通常选用g-球蛋白为标准蛋白质以减少该方面的误差;仍存在一些物质干扰此方法的测定,如去污剂、Triton X-100、十二烷基硫酸钠(SDS)和0. 1N的NaOH等;标准曲线也有轻微的非线性(尤其是在溶液浓度较高时),故不能用朗伯-比尔定律进行计算而只能用标准曲线来确定未知蛋白质的浓度。
2.实验可行性分析
就实验方案而言,本实验首先通过对不同浓度标准蛋白溶液的测定与误差分析来确定不同测定方法的灵敏度及准确性,然后测定生物的未知蛋白粗提取物中蛋白质含量,分别做出不同方法所得到测定值的两个波动趋势,分析他们之间是否具有相似性,从而得出非蛋白组分的影响在蛋白质含量测定中是否可以忽略。方案合理,简便易行,具有可行性。另外,实验中所要用到的材料、试剂、仪器在生物化学实验室中均可方便获得,故就实验器具而言也具有可行性。下文中会通过具体实验流程计算该实验大概所需时间为5h,时间上也具有可行性。综合以上各点,该设计实验具有可操作性。
3.预期的实验结果
(1)高浓度时紫外法测定的蛋白质含量最接近真实值,低浓度时考马斯亮蓝(G-250)法测定的蛋白质含量最接近真实值;
(2)非蛋白组分在蛋白质含量测定中确实存在着影响,且该影响在实际测定过程中不能够忽略。
五、具体实验设计
1.主要实验材料、试剂及仪器
紫外分光光度计( )、722型分光光度计( )、分析天平( )、移液管( )、试管( )、具塞刻度试管( )、50ml容量瓶( )、100ml容量瓶( )、研钵( )、乙醇( )、标准牛血清白蛋白溶液(1mg/ml BSA,250μg/ml BSA,100μg/ml BSA)、待测样液1(待测液1A,500μg/ml BSA、待测液1B,250μg/ml BSA、待测液1C,100μg/ml BSA、待测液1D,10μg/ml BSA)、待测样液2(生物样品的蛋白质粗提取液)、Folin-酚试剂甲、Folin-酚试剂乙、考马斯亮蓝G-250。
2.操作步骤
2.1 紫外法测定蛋白质含量
实验流程图:
牛血清白蛋白标准溶液(1 mg/ml)的配制(配方见附录1)

牛血清白蛋白标准溶液(1 mg/ml)与蒸馏水混合得到0-1 mg/ml蛋白质溶液
0-1 mg/ml蛋白质溶液光密度值OD280的测定(用光程1cm的石英比色杯)
绘制0-1 mg/ml标准曲线
待测液1(四组1A、1B、1C、1D)OD280的测定(每组测3次取平均值)
待测液2 OD280的测定(测3次取平均值)
蛋白质含量的计算
原始数据记录处:
表1 0-1 mg/ml蛋白质溶液OD280值
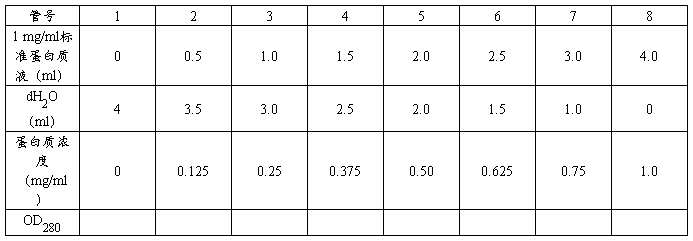
表2 待测液1的OD280值及蛋白质浓度

表3 待测液2的OD280值及蛋白质浓度

预计用时:40min
2.2 Folin-酚法(Lowry法)测定蛋白质含量
实验流程图:
牛血清白蛋白标准溶液(250μg/ml)的配制(配方见附录1)
牛血清白蛋白标准溶液(250μg/ml)与蒸馏水混合得到0-250μg/ml蛋白质溶液
分别加入5ml试剂甲,室温放置10min
分别加入0.5ml试剂乙混匀,放置半小时
0-250μg/ml蛋白质溶液吸光度值A650的测定
绘制0-250μg/ml标准曲线
待测液1(四组1A、1B、1C、1D)A650的测定(方法同上,每组测3次取平均值)
待测液2 A650的测定(方法同上,测3次取平均值)
蛋白质含量的计算
原始数据记录处:
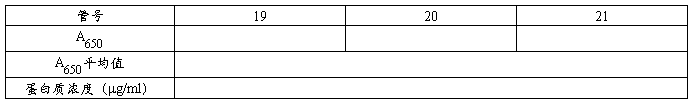
表4 0-250μg/ml蛋白质溶液A650值

表5 待测液1的A650值及蛋白质浓度

表6 待测液2的A650值及蛋白质浓度
预计用时:2h
2.3 考马斯亮蓝(G-250)法(Bradfold法)
实验流程图:
牛血清白蛋白标准溶液(100μg/ml)的配制(配方见附录1)
牛血清白蛋白标准溶液(100μg/ml)与蒸馏水混合得到0-100μg/ml蛋白质溶液
分别加入5ml考马斯亮蓝G-250,混匀,放置两分钟(10ml具塞试管)
0-100μg/ml蛋白质溶液吸光度值A595的测定
绘制0-100μg/ml标准曲线
绘制0-1000μg/ml标准曲线(方法同上)
待测液1(四组1A、1B、1C、1D)A595的测定(方法同上,每组测3次取平均值)
待测液2 A595的测定(方法同上,测3次取平均值)
蛋白质含量的计算
原始数据记录处:
表7 0-100μg/ml蛋白质溶液A595值
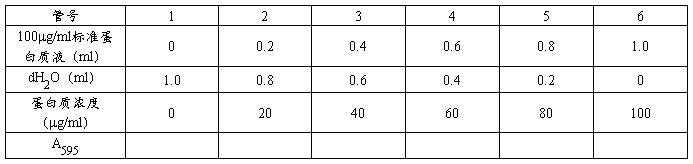
表8 0-1000μg/ml蛋白质溶液A595值
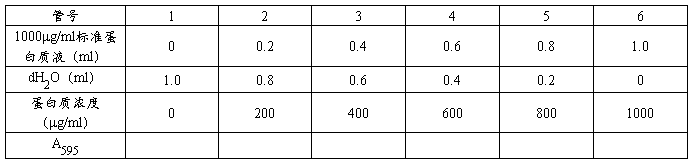
表9 待测液1的A595值及蛋白质浓度
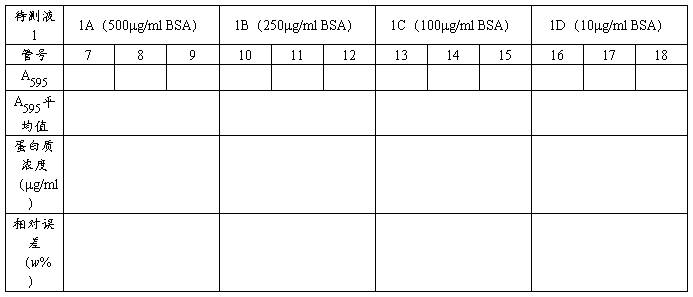
表10 待测液2的A595值及蛋白质浓度

预计用时:50min
六、质疑与思考
(1)本实验使用的三种蛋白质测定方法其原理都是基于分光光度法,该方法与其他蛋白质测定法相比有什么优点?使用该方法时都应该注意什么?
分光光度法测量浓度的基本原理是朗伯-比尔定律(A=εbc),当固定溶液层厚度b和吸光系数ε时,吸光度A与溶液的浓度c成线性关系。该方法具有明显的优点,即灵敏度高(常用于测定试样中的为微量组分)、精准度高(相对误差在2%-5%)、操作简便快捷、适用范围广(大多数无机离子和有机化合物都可通过分光光度法直接或间接测定)。
使用该方法时应注意:1需要选定入射光的波长。选择被测物质的最大吸收波长作为入射光波长,这样测量时灵敏度最高,偏离朗伯-比尔定律的程度越小。2要控制适当的吸光度范围,一般应控制标准溶液和待测样液的吸光度在0.15-0.8范围内。3选择适当的参比溶液(空白对照)。
(2)在绘制标准曲线时,空白组的设定是不是必须的?
空白组溶液是用来调节仪器工作零点的,当不设置空白组或者空白组选取不适当时都会对测量读数的准确度有较大的影响。空白组的选取遵循以下几个原则:1纯溶剂空白:当试液、试剂、显色剂均在测定波长处无吸收时,可用蒸馏水作参比液,称纯溶剂空白。2试液空白:试剂和显色剂均无色时,而被测溶液中其他离子有色时,应采用不加显色剂的试液溶液作参比液,称试液空白。3试剂空白:试剂和显色剂均有颜色时,可将一份试液加入适当掩蔽剂,将被测组分掩蔽起来,使之不再与显色剂作用,然后把显色剂、试剂均按试液测定方法加入,以此做参比溶液,称试剂空白。
(3)通过比较曲线的变化趋势来确定非蛋白组分对蛋白质含量测定产生的干扰程度这一方法存在一定的不科学性
在本实验中,以牛血清白蛋白为标准蛋白来绘制标准曲线,待测样液1各组(1A、1B、1C、1D)所含的蛋白质也是牛血清白蛋白,而待测样液2(生物样品的蛋白质粗提取液)中的蛋白质种类未知。如果待测样液2中的蛋白质与牛血清白蛋白中的酪氨酸、色氨酸残基含量差异较大,则三种方法测得的样液2中蛋白质含量会有较大的偏差,造成最终获得的两条蛋白质含量曲线图之间没有可比性,也就不能够判断非蛋白组分对蛋白质含量测定产生的干扰有多大。
(4)该实验仅仅通过测定一种生物样品的蛋白质提取液,就推断非蛋白组分的干扰在蛋白质含量测定中能否忽略,这是缺乏说服力的。因为在同一生物体的不同组织中蛋白质含量存在差异,在不同生物体之间就更不一样,且非蛋白组分的种类也不同,它们有的对蛋白质含量的测定干扰很小,有的则干扰很大。所以本实验最终的结果并不能推而广之。
(5)实验误差分析
实验中容易产生误差的地方:(1)标准蛋白质溶液的的配制。在溶液配制过程中应该严格按照操作流程进行准确称量、少量多次洗涤、准确定容,以保证配出的标准蛋白溶液符合要求。(2)Folin-酚法测定蛋白质含量时时间的控制。该方法测定时,要严格控制时间,加入甲液后放置10分钟,加入乙液后混匀放置半小时,以便呈现出最稳定的颜色。
七、注意事项
(1)正确使用移液器(枪);
(2)试管使用前必须清洗干净,并干燥;
(3)比色杯以及玻璃器皿对考马斯亮蓝G-250吸附非常厉害,使用后应先用乙醇清洗。
八、参考文献
[1]李宁.几种蛋白质测定方法的比较[J].山西农业大学学报,2006,02(03):132-134.
[2]魏琴等.蛋白质分析技术的发展趋势[J].济南大学学报(自然科学版),2003,4(17):312-320.
预习提问
1.当你想从一个混合的生物体系中提取某一物质时,你的整体思路是怎样的?(即:你如何去实现这一目标?),也就是物质分离纯化的总体战略是什么?
个人认为在生物体系中分离提纯某一物质时需要遵循以下几个原则:操作要简便易行(步骤尽可能少)、速度快;分离提纯过程中所消耗的材料、设备以及能源等各种费用要尽可能少;分离提纯所得到的目标产物的回收率要尽可能高;目标产物的纯度也要尽可能的高;不能改变目标产物的活性以及空间结构。基于以上原则,在生物体系中分离纯化某一物质时,首先需要了解要分离的物质具有哪些特点,利用目标物质与杂质具有显著差异的特性,如溶解度、分子大小、电荷性质、吸附性等,采用合适的物理化学方法,使目标物质与杂质得到分离。
2.根据上学期所学的知识总结一下生物大分子比之以往的化学物质其特点是什么?针对这些特点在进行生物大分子的分离纯化时应该注意什么?
生物大分子的特点:(1)是相对分子质量很大的有机物;(2)一般都由特定的基本结构单元聚合而成,如蛋白质的基本单位是氨基酸;(3)空间结构复杂,如蛋白质、核酸等都具有多级结构;(4)多数生物大分子具有活性,能够维持特定的生理功能;(5)次级键(弱键)对于稳定与维持生物大分子的结构和功能有着重要的作用,另外生物大分子结构和功能的维持需要适宜的外界条件,如温度、pH等;(6)生物大分子本身具有一些物理特性,诸如溶解度、吸附性等;(7)生物大分子在生物体内能够降解和再生。
基于以上特点,在进行生物大分子的分离纯化时为避免其变性、降解,需要在合适的条件下进行操作(低温、使用缓冲液),另外要加入抗氧化还原的物质或者蛋白酶抑制剂避免其发生氧化还原反应及被酶分解,总之,不能够破坏其结构与活性。
3.在生物化学实验研究中,为什么要使用缓冲液?在使用缓冲液时应该注意哪些问题?
(1)使用缓冲液的原因是生物体内发生的各种化学反应都需要在适宜的pH下才能够正常进行,一些生物大分子(如酶等)也需要在适宜的pH下才具有生物活性,而不同的缓冲液则能够将pH稳定在特定的范围内,为生化反应提供适宜的外界条件。
(2)使用缓冲液时应该注意:要根据条件需要的pH范围选择适宜的缓冲液;要考虑缓冲液的成分是否对实验产生干扰(如NH4+、PO43-等)。
附录
1.标准牛血清白蛋白溶液的配制(预计时间20min)
在分析天平上准确称取100mg结晶牛血清白蛋白(BSA),倒至小烧杯中,加入少量蒸馏水之后转入100ml容量瓶中,烧杯内的残液用蒸馏水少量多次清洗,并将冲洗液一并转入容量瓶中,最后用蒸馏水定容至刻度,配制成标准原液,其中牛血清白蛋白的浓度为1mg/ml(1mg/ml BSA)。浓度为250μg/ml的标准牛血清白蛋白溶液(250μg/ml BSA)配制时称取BSA 25mg,100μg/ml的标准牛血清白蛋白溶液(100μg/ml BSA)配制时称取BSA 10mg,其余步骤同上。
2.待测样液1的配制(预计时间20min)
待测样液1A(500μg/ml BSA):配制时称取BSA 50mg,其余步骤同附录1;
待测样液1B(250μg/ml BSA):配制方法见附录1;
待测样液1C(100μg/ml BSA):配制方法见附录1;
待测样液1D(10μg/ml BSA):配制时称取BSA 1mg,其余步骤同附录1;
3.待测样液2的配制(预计时间1h)
称取新鲜的绿豆芽下胚轴3g放入研钵中,滴加适当蒸馏水研磨成匀浆,将匀浆转入离心管中,再用适量蒸馏水少量多次洗涤研钵,洗涤液转入同一离心管中,搅拌半分钟,室温下放置0.5-1h以充分提取,然后在4000r/min下离心20min,弃去沉淀物,将上清液转入容量瓶,随后用蒸馏水定容至50ml,该溶液即为待测样液2。
4.Folin-酚试剂的配制
(1)Folin-酚试剂甲:
A液:10gNa2CO3、2gNaOH和0.5克酒石酸钾钠 (KNaC4H4O6?4H2O)溶解后用蒸馏水定容至500ml。
B液:0.5g硫酸铜(CuSO4?5H2O)溶解于100ml蒸馏水中,每次使用前将50份A液与1份B液混合,即为试剂甲。
(2)Folin-酚试剂乙
在1.5L磨口回流瓶中加入100g钨酸钠(Na2WO4?2H2O)和25克钼酸钠(Na2MoO4?2H2O)及700ml蒸馏水,再加50ml85%磷酸、100ml浓盐酸,充分混合,接上回流管,以小火回流10小时。回流结束后,加入150g硫酸锂(Li2SO4),50ml蒸馏水及数滴液体溴,开口继续沸腾15分钟,以便驱除过量的溴,冷却后溶液呈黄色(如仍呈绿色,须再重复滴加液体溴的步骤)。稀释至1L,过滤,滤液置于棕色试剂瓶中保存。使用时用标准NaOH滴定,酚酞作指示剂,然后适当稀释,约加水1倍,使最终的酸浓度为1N左右。
5.考马斯亮蓝G-250的配制
称取100mg考马斯亮蓝G-250,溶于50ml90%的乙醇中,加入体积分数为85%的磷酸100ml,最后用蒸馏水定容到1000ml,此溶液在常温下可放置一个月。
实验记录处
-
蛋白质的定量测定实验报告
显问题其次回顾全部过程的原理我们猜测可能存在的造成结果低的情况为在室温冷却后实验室没有空余的分光光度计排队时间应该是全部小组中最长…
-
蛋白质测定实验报告
蛋白质测定方法化学报告蛋白质的检测酚试剂法灵敏度较高20250mg费时蛋白质在碱性溶酚类柠檬液中其肽键与酸硫酸铵Cu2螯合形成tr…
-
蛋白质测定实验报告
生物化学实验报告姓名学号专业年级组别生物化学与分子生物学实验教学中心实验名称实验日期20xx1015合作者评分Folin酚试剂法测…
-
紫外分光光度法测定蛋白质含量实验报告
紫外分光光度法测定蛋白质含量一实验目的1学习紫外光度法测定蛋白质含量的原理2掌握紫外分光光度法测蛋白质含量的实验技术二实验原理1测…
-
分光法测蛋白质实验报告
紫外分光光度法测定蛋白质含量一实验目的1学习紫外分光光度法测定蛋白质含量的原理2掌握紫外分光光度法测定蛋白质含量的实验技术3掌握T…
-
蛋白质测定实验报告
蛋白质测定方法化学报告蛋白质的检测酚试剂法灵敏度较高20250mg费时蛋白质在碱性溶酚类柠檬液中其肽键与酸硫酸铵Cu2螯合形成tr…
-
蛋白质功能性质的检测实验报告
华南农业大学实验报告专业班次组别题目蛋白质功能性质的检测姓名黄俊怡日期20xx0711一实验目的通过本实验定性地了解蛋白质的主要功…
-
SCAU实验报告蛋白质功能性质的检测
蛋白质功能性质的检测一实验目的通过本实验定性地了解蛋白质的主要功能性质二实验原理蛋白质的功能性质一般是指能使蛋白质成为人们所需要的…
-
实验报告 蛋白质分子的测定
实验一蛋白质分子的测定凝胶层析法一实验原理凝胶层析法即凝胶过滤法gelfiltration是利用凝胶把分子大小不同的物质分离开的一…
-
蛋白质的定量测定实验报告
显问题其次回顾全部过程的原理我们猜测可能存在的造成结果低的情况为在室温冷却后实验室没有空余的分光光度计排队时间应该是全部小组中最长…
-
生化实验报告 福林-酚试剂法测定蛋白质的浓度
实验三福林Folin酚试剂法测定蛋白质的浓度一实验目的1学会制备无蛋白血滤液2掌握FolinWu法测定血糖含量的原理和方法3掌握7…