药学系本科生毕业论文格式模版
目 录
一、开题报告……………………………………………………………………………1 1 课题名称………………………………………………………………………… 1 2 课题来源………………………………………………………………………… 1 3 立题依据………………………………………………………………………… 1 4 研究内容、研究目的和拟解决的关键问题…………………………………… 2 5 拟采用的实验方法、步骤、技术路线及可行性分析………………………… 2 6 研究计划及预期进展…………………………………………………………… 2 7 预期研究结果…………………………………………………………………… 2
二、毕业论文……………………………………………………………………………3 1 作者……………………………………………………………………………… 3 2 指导老师………………………………………………………………………… 3 3 中英文摘要……………………………………………………………………… 3 4 关键词…………………………………………………………………………… 3 5 正文……………………………………………………………………………… 4 6 参考文献………………………………………………………………………… 12 7 致谢……………………………………………………………………………… 13
三、综述…………………………………………………………………………………14 1 摘要……………………………………………………………………………… 14 2 关键词…………………………………………………………………………… 14 3 正文……………………………………………………………………………… 14 4 参考文献………………………………………………………………………… 16
四、译文…………………………………………………………………………………18
开题报告
1 课题名称:RS有机溶剂残留量测定
龙 伟 药学系药学班 20027013
指导老师 廖一静 药物分析教研室
黄荣清 军事医学科学院 副研究员
肖炳坤 军事医学科学院 助理研究员
2 课题来源:军事医学科学院基金
3 立题依据:
3.1 研究意义:药物中的残留溶剂是指在药物的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物。由于残留溶剂不仅没有疗效,还可能增加药物的毒副作用,影响药物的稳定性,而且溶剂挥发容易造成环境的污染。出于对患者的安全和对环境的保护,所有的有机溶剂应尽可能除去,药品注册时对有机溶剂残留量有相应的要求,需对药品在生产过程中引入的有机溶剂残留量进行测定,RS是一个经过多步化学反应得到的一种药物,在合成过程中使用到的一些有机溶剂必须尽可能的除去,使之符合药物质量标准。
3.2 国内外研究现状:国内外目前对去有机溶剂残留量的测定有多种方法可以选择,一般选用的都是色谱法,而毛细管GC法由于其检测灵敏度高,塔板数高,分离效果好等优点,成为当前一种比较实用,常见,方便的方法。
3.3 存在的问题:由于在合成过程中,使用到的三中有机溶剂分别为甲醇,乙酸,DMF,他们之间的性质是存在比较大的差异的,所以无法同时测定,所以我们采用分别测定的方法;由于DMF的沸点比较高,所以在要摸索合适的梯度温度是实验要解决的一个重要问题。
3.4 参考文献:
[1] 吴烈钧.气相色谱检测方法.傅若农主编.色谱技术丛书[M]. 北京:化学工业出版社,2000.
[2] 美国瓦里安中国有限公司分析仪器部.[J].国外分析仪器.1996;(3):85.
[3] 许国旺、叶纷等.色谱,2001,19(2):132-135
[4] LI HUI-yi(李惠义),WEI jing-jing(魏京京),ZHU Wei-hua(朱维华).GC Detection of Residual Solvents in Iguratimod(艾垃谟德中残留溶剂的检测方法的研究).Chin J Pharm
1
Anal,2004,24(4):422
[5] 骆传环,黄荣清,邢成,等. 沙纳唑含量测定及体外放射增敏作用。中华放射医学与防护杂志.
[6] Bull RJ,Health.Hazard associate with organic chemical in drinking water Application of short-term biassays in the analysis ofcomplexenviromental mixture,1981,(2)
4 研究内容、研究目的和拟解决的关键问题
4.1 研究内容:主要包括对RS有机溶剂残留量测定方法的摸索,溶剂的选择,温度梯度的摸索,通过对条件的摸索,建立在方法学上可以确证的,RS有机溶剂残留量测定方法
4.2 研究目的:在药典规定的框架下建立一套对RS中有机溶剂残留量测定的有效,方便的方法。
4.3 拟解决的关键问题:首先需要解决对有机溶剂溶媒的适当选择,其次需要对其温度梯度的选择,线性的拟合。
5 拟采用的实验方法、步骤、技术路线及可行性分析
5.1 实验方法、步骤、技术路线:本试验采用常用有机溶剂残留量测定方法,即气相外标法定量,选用高纯度样品,用高效液相进行样品纯度检测。选择适当的溶解介质和温度梯度,作出标准曲线,然后测定样品,同时考察精密度,系统适应性测试,最低检测限,最低定量限等作为方法学确证的依据,回归方程外标法计算结果。进样方法为手动进样。
5.2 可行性分析:本试验所用方法,是当前国内外有机溶剂残留量测定方法中较为常用的方法,是按照药典的各项要求进行,所用样品经高效液相检测纯度,各项试剂,仪器均符合标准,是切实可行的方案。
6 研究计划及预期进展:本课题计划在三个月内完成,预计三月份完成其样品溶解介质的筛选,对样品纯度进行测试,四五月份完成对其合适分离条件如温度梯度的选择的选择。对各个样品进行GC检测。
7 预期研究结果:建立一套尽可能简便,快捷,有效的RS中有机溶剂残留量GC测定方法,作为质量研究的一部分。为新药的申报注册补充资料。
指导老师: 黄荣清 肖炳坤 廖一静 20xx年4月 10 日
2
毛细管气相色谱法测定RS中有机溶剂残留量
作 者:龙 伟 药学系02级药学班
指导老师:廖一静 药物分析教研室
黄荣清 军事医学科学院 副研究员
肖炳坤 军事医学科学院 助理研究员
摘要:目的:建立RS中有机溶剂的残留量的测定方法。方法:采用毛细管气相色谱法,色谱柱为DB-WAX(30m×0.45mm×0.85μm)石英毛细管柱;检测器为FID,载气为氮气;柱温为程序升温:起始温度为80℃,然后以30℃/min的速率升至150℃,再以10℃/min的速率升至200℃,保持7 min;气化室温度为210℃;外标法计算含量。结果:在该色谱条件下,测得甲醇,乙酸,DMF三种溶剂分别在4.0~63.2 μg/ml,21~210μg/ml,和
4.8~47.5μg/ml范围内线性关系良好(r=0.9998,0.9998和0.9982);平均回收率分别为90.8%,109.6%和105.0%,RSD分别为2.9%,1.4%和3.8%;最低检测限分别为0.4ng,
6.3ng和0.95ng。结论:该毛细管气相色谱法准确、灵敏、可靠,适用于本品有机溶剂残留量的测定,测定结果符合药典规定要求。
关键词 RS;有机溶剂残留量;毛细管气相色谱法
The Residual Solvents Detection of RS by Capillary GC
Long Wei (Class Pharmacy, Grade 2002 in Department of Pharmaceutical Science,
Nanchang University Medical College)
Derector: Liao Yi jing (Teaching and Research Section of Pharmaceutical Analysis) Huang Rong-qing (Military Medical academy of sciences, associate researcher )
ABSTRACT Objective:To establish a method for determination of residual organic solvents in RS. Methods:A capillary GC method was developed with
DB-Wax(30m×0.45mm×0.85μm) quartzose capillary column with FID detector and nitrogen as the carries gas; the column temperature rose by program, the initial temperature was 80℃, the temperature was raised to 150℃ at the rate of 30℃/min, then temperature was raised to 200℃ at the rate of 10℃/min, maintain for 7 min ,the injector temperature was 210℃;
3
calculate the content by external standard method . Result:The analytical balidation for each of residual organic solvents was performed, The linearities were good between 4.0~63.2 μg/ml,21~210μg/ml,and 4.8~47.5μg/ml of methanol, acetic acid and DMF respectively (r=0.9998,0.9998and 0.9982); the average recoveries were 90.8%,109.6%,and 105.0% respectively with RSD of 2.9%,1.4% and 3.8%; the limit of detection were 0.4ng,6.3ng and 0.95ng. Conclusions: This GC method is proved to be accurate, sensitive, reliable and suitable for the residual solvents detection of RS. The results is accord with the request of pharmacopeia.
Key words RS; the contents of residual organic solvents; Gas Chromatography(GC)
药物中的残留溶剂是指在原料药或赋型剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物。由于残留溶剂不仅没有疗效,还可能增加药物的毒副作用,而且影响药物的稳定性,故所有的有机溶剂应尽可能除去。为了保护患者免受药物中残留有机溶剂的伤害,需对药品在生产过程中引入的有机溶剂残留量进行测定,RS是一个经过多步化学反应得到的药物,是一种放射增敏剂,它有助于解决肿瘤组织中缺乏氧细胞对射线的抗性而导致放疗的治愈率低的问题,不仅有杀死肿瘤细胞的作用,而且对化疗有很好的促进作用,有着良好的应用前景。在它的有机合成过程中使用了有机溶剂甲醇,乙酸,N,N-二甲基甲酰胺(DMF),根据国家药物申报要求,必须对其最后成品中的有机溶剂残留量进行检查。本文建立了RS中甲醇,乙酸,DMF残留量的GC检查方法,同时测定了RS中甲醇,乙酸,DMF残留量,结果表明本法分离度高,灵敏,准确而且方便。可以用于本品有机溶剂残留量的检测。
1 材料
1.1 药品与试剂
RS 021110(99.84%,军事医学科学院);RS 030315(99.75%,军事医学科学院) RS 030321(99.84%,军事医学科学院);二甲基亚砜(AR,北京化工厂);甲醇(AR, 北京化工厂);乙酸(AR,北京化工厂);N,N-二甲基甲酰胺(DMF)(AR,北京化工厂)。
1.2 仪器
美国Agilent公司6820型气相色谱仪;FID检测器。
4
2 色谱条件与系统适应性试验
2.1 色谱条件
色谱柱:为DB-WAX(固定相为聚乙二醇)30m×0.45mm×0.85μm;气化室温度为210℃;检测器温度为250℃;载气为高纯氮气,柱温为程序升温:起始温度为80℃,然后以30℃/min的速率升至150℃,再以10℃/min的速率升至200℃,保持7 min。流速为2.6ml/min,柱头压力为3.5psi,分流比为14:1,空气流速为260ml/min,氢气流速为37ml/ml,辅助气流速为24ml/min,进样量1μl。
2.2 系统适应性试验
在上述气相色谱条件下,取甲醇、乙酸、N,N-二甲基甲酰胺(DMF)适量,用二甲基亚砜稀释进样,记录气相色谱;同法记录空白溶剂(二甲基亚砜)的气相色谱图;由图可知溶剂对残留有机溶剂的测定没有干扰。在此气相色谱图条件下,甲醇、乙酸、N,N-二甲基甲酰胺能达到完全分离,色谱柱理论塔板数按各峰计算均大于1000。(图谱见24页)
3 对照品溶液的配制与标准曲线的制备
3.1 对照品溶液的配制
精密量取甲醇5μl、乙酸20μl和N,N-二甲基甲酰胺(DMF)5μl分别置于3个10ml量瓶中,加二甲基亚砜(DMSO)稀释至刻度,摇匀,作为对照品贮备液;再从中分别精密量取0.4ml置于另3个10ml量瓶中,加二甲基亚砜稀释至刻度,摇匀,作为对照品溶液。精密量取对照品溶液1μl,注入气相色谱仪,记录色谱图
3.2 标准曲线的的制备
3.2.1 甲醇标准曲线制备
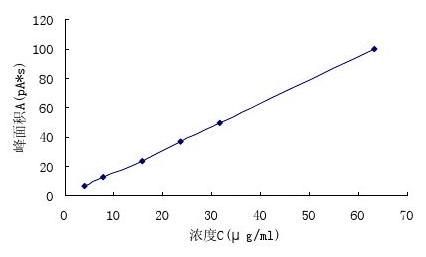
分别精密量取甲醇对照品贮备液各0.1、0.2、0.4、0.6、0.8、1.6ml置10ml量瓶中,加二甲基亚砜稀释至刻度,摇匀。精密量取各溶液1μl,注入气相色谱仪,记录色谱图,以各溶液浓度C(μg/ml)为横坐标,峰面积A(pA*s)为纵坐标进行线性回归,结果见表1。
表1 甲醇的浓度(C)与峰面积(A)关系
Tab. 1 The relationship of methol concetration and peak area
浓度C(μg/ml)
峰面积A(pA*s) 4.0 6.4 7.9 12.8 15.8 23.6 23.7 36.8 31.6 50.0 63.2 100.3
5
图1甲醇的浓度(C)与峰面积(A)的线性图
Fig.1 Linear graph of methanol concentration and peak area
回归方程:A=1.58992C-0.42435;R=0.99982。
结果表明:甲醇在4.0~63.2 μg/ml范围内呈良好的线性关系。 3.2.2 乙酸标准曲线制备
分别精密量取乙酸对照品贮备液各0.1、0.2、0.4、0.6、0.8、1.0ml置10ml量瓶中,加二甲基亚砜稀释至刻度,摇匀。精密量取各溶液1μl,注入气相色谱仪,记录色谱图,以各溶液浓度C(μg/ml)为横坐标,峰面积A(pA*s)为纵坐标进行线性回归,结果见表2。
表2 乙酸的浓度(C)与峰面积(A)关系 Tab. 2 The relationship of acetate concetration and peak area
浓度C(μg/ml) 峰面积A(pA*s)
21 9.5
42 20.7
84 41.5
126 62.8
168 82.8
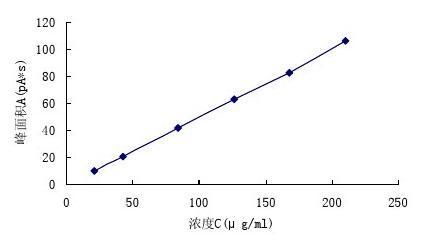
210 106.1
图2 乙酸的浓度(C)与峰面积(A)的线性图
Fig.2 Linear graph of acetic acid concentration and peak area
6
回归方程:A=0.49861 C-0.51707;R=0.9998
结果表明:乙酸在21~210μg/ml范围内呈良好的线性关系。
3.2.3 N,N-二甲基甲酰胺标准曲线制备
分别精密量取N,N-二甲基甲酰胺对照品贮备液各0.1、0.2、0.4、0.6、0.8、1.0ml置10ml量瓶中,加二甲基亚砜稀释至刻度,摇匀。精密量取各溶液1μl,注入气相色谱仪,记录色谱图,以各溶液浓度C(μg/ml)为横坐标,峰面积A(pA*s)为纵坐标进行线性回归,结果见表3。
表3 N,N-二甲基甲酰胺的浓度(C)与峰面积(A)关系
Tab. 3 The relationship of DMF concetration and peak area
浓度C(μg/ml)
峰面积A(pA*s)
4.8 5.7 9.5 12.0 19.0 21.9 28.5 33.9 38.0 45.7
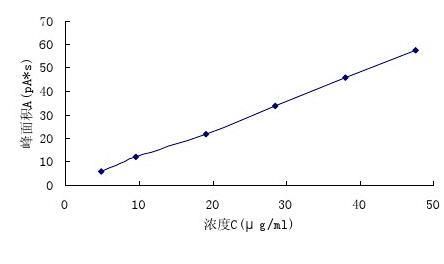
47.5 57.5
图3 DMF的浓度(C)与峰面积(A)的线性图
Fig.3 Linear graph of DMF concentration and peak area
回归方程:A=1.18362C+1.02088;R=0.9982。
结果表明:N,N-二甲基甲酰胺在4.8~47.5μg/ml范围内呈良好的线性关系。
4 精密度试验
4.1 甲醇精密度实验
将甲醇对照品溶液连续重复进样6次,各1μl,记录峰面积,并计算其峰面积的RSD(%)。结果6次的峰面积(pA*s)分别为:24.1,23.0,21.7,21.2,22.1,21.2,RSD=5.1%。
4.2 乙酸精密度实验
将乙酸对照品溶液连续重复进样6次,各1μl,记录峰面积,并计算其峰面积的RSD(%)。结果6次的峰面积(pA*s)分别为:41.4,43.6,41.6,41.3,42.6,42.8,RSD=2.19%。
7
4.3 N,N-二甲基甲酰胺精密度实验
将N,N-二甲基甲酰胺对照品溶液连续重复进样6次,各1μl,记录峰面积,并计算其峰面积的RSD(%)。结果5次的峰面积(pA*s)分别为:22.0,21.7,22.8,21.6,21.7,22.1,RSD=2.02%。
5 最低检测限与最低定量限试验 5.1 最低检测限试验
将甲醇,乙酸和N,N-二甲基甲酰胺分别用二甲基亚砜逐步稀释,直到检测峰高为基线噪音的2~3倍为止,结果三者的最低检测限(ng)分别为0.40, 2.1, 0.95。 5.2 最低定量限试验
将甲醇, 乙酸和N,N-二甲基甲酰胺用二甲基亚砜逐步稀释,直到检测峰高为基线噪音的10倍为止,结果三者的最低定量限(ng)分别为为1.3,6.3,2.9。 6 回收率试验 6.1 甲醇回收率实验
取样品(批号021110)300mg,精密称定,置10ml量瓶中,加甲醇对照品溶液溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪进行测定;另取样品(批号021110)300mg,精密称定,置10ml量瓶中,加二甲基亚砜溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪测定甲醇残留量,同时取1μl甲醇对照品溶液注入气相色谱仪进行测定,平均峰面积为20.9;计算出回收率结果见表4。
表4 甲醇回收率实验结果 Tab. 4 The recovery result of methol
样品量 Area(pA*s) 11.8 11.8 11.8 11.8 11.8 11.8
标准加入量 Area(pA*s) 20.9 20.9 20.9 20.9 20.9 20.9
样品+对照品
实测加入量
回收率 (%) 88.0 91.4 90.0 90.0 95.7 89.5 90.8 2.9
Area(pA*s) Area(pA*s) 30.2 30.9 30.6 30.6 31.8 30.5
18.4 19.1 18.8 18.8 20.0 18.7
平均回收率(%)
RSD(%)
8
6.2 乙酸回收率实验
取样品(批号021110)300mg,精密称定,置10ml量瓶中,加N,N-二甲基甲酰胺对照品溶液溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪进行测定;另取样品(批号021110)300mg,精密称定,置10ml量瓶中,加二甲基亚砜溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪测定N,N-二甲基甲酰胺残留量;同时取1μl N,N-二甲基甲酰胺对照品溶液注入气相色谱仪进行测定,平均峰面积为23.8;计算出回收率结果见表5。
表5 N,N-二甲基甲酰胺回收率实验结果 Tab.5 The recovery result of acetate
样品量 Area(pA*s)
0 0 0 0 0 0
标准加入量 Area(pA*s) 23.8 23.8 23.8 23.8 23.8 23.8
样品+对照品
实测加入量
回收率 (%) 100.8 105.5 112.2 102.1 103.8 105.5 105.0 3.8
Area(pA*s) Area(pA*s) 24.0 25.1 26.7 24.3 24.7 25.1
24.0 25.1 26.7 24.3 24.7 25.1
平均回收率(%)
RSD(%)
6.3 N,N-二甲基甲酰胺回收率实验
取样品(批号021110)300mg,精密称定,置10ml量瓶中,加N,N-二甲基甲酰胺对照品溶液溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪进行测定;另取样品(批号021110)300mg,精密称定,置10ml量瓶中,加二甲基亚砜溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪测定N,N-二甲基甲酰胺残留量;同时取1μl N,N-二甲基甲酰胺对照品溶液注入气相色谱仪进行测定,平均峰面积为23.8;计算出回收率结果见表6。
9
表6 N,N-二甲基甲酰胺回收率实验结果
Tab. 6 The recovery result of DMF
样品量 Area(pA*s)
0 0 0 0 0 0
标准加入量 Area(pA*s) 23.8 23.8 23.8 23.8 23.8 23.8
样品+对照品
实测加入量
回收率 (%) 100.8 105.5 112.2 102.1 103.8 105.5 105.0 3.8
Area(pA*s) Area(pA*s) 24.0 25.1 26.7 24.3 24.7 25.1
24.0 25.1 26.7 24.3 24.7 25.1
平均回收率(%)
RSD(%)
7 样品测定 7.1 甲醇残留量测定
取3批样品(批号021110、030315、030321)各300mg,精密称定,置10ml量瓶中,加二甲基亚砜溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪测定甲醇残留量;同时取1μl甲醇对照品溶液进行测定,结果见表7。
表7 3批样品中甲醇残留量测定结果 Tab. 7 The methol residual result of 3 samples
甲醇残留量
批号
峰面积(pA*s) 实测值
021110 030315 030321
11.5 12.4 12.0
12.0 12.7 12.2
平均值 11.8 12.6 12.1
浓度 (%) 0.03 0.03 0.03
0.3 药典规定的 浓度限度(%)
7.2 乙酸残留量测定
取3批样品(批号021110、030315、030321)各300mg,精密称定,置10ml量瓶中,加二甲基亚砜溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪测定乙酸残留量;同时取1μl乙酸对照品溶液进行测定,结果见表8。
10
表8 3批样品中乙酸残留量测定结果 Table 8 The acetate residual result of 3 samples
乙酸残留量
批号
峰面积(pA*s) 实测值
021110 030315 030321
3.4 7.6 8.2
3.3 7.3 7.9
平均值 3.4 7.5 8.1
浓度 (%) 0.024 0.053 0.057
0.5 药典规定的 浓度限度(%)
7.3 N,N-二甲基甲酰胺残留量测定
取3批样品(批号021110、030315、030321)各300mg,精密称定,置10ml量瓶中,加二甲基亚砜溶解并稀释至刻度,摇匀,取1μl注入气相色谱仪测定乙酸残留量;同时取1μlN,N-二甲基甲酰胺对照品溶液进行测定,结果见表9。
表9 3批样品中N,N-二甲基甲酰胺残留量测定结果
Tab. 9 The DMF residual result of 3 samples
N,N-二甲基甲酰胺残留量
批号
峰面积(pA*s) 实测值
021110 030315 030321
0 0 0
0 0 0
平均值 0 0 0
浓度 (%) 0 0 0
0.088 药典规定的 浓度限度(%)
8.结果与讨论 8.1 实验结果
通过测定,在RS制备过程中,使用过的三种有机溶剂甲醇、乙酸、N,N-二甲基甲酰胺残留量结果见表10。可以与药典标准进行比较。
表10 3批样品中各溶剂残留量测定结果 Tab. 10 The solvent residual result of 3 samples
样品批号 甲醇(%) 乙酸(%) N,N-二甲基甲酰胺(%)
021110 0.03 0.024 0
030315 0.03 0.053 0
11
030321 0.03 0.057 0
药典规定的浓度限度
0.3 0.5 0.088
从表中数据我们可以看出,原料药RS中甲醇、乙酸、N,N-二甲基甲酰胺残留量远低于药典所规定的标准值,符合药典要求。
8.2 讨论
在色谱柱的选择上,由于毛细管柱法的分离度好,灵敏度高,因此本法采用毛细管柱法,测定甲醇,乙酸,DMF残留量,效果令人满意。
一般来说,在有机溶剂残留量的测定是可以用水来溶解(FID检测器),但由于本品难溶于水,故采用DMSO溶解,DMSO对各个待测物均有良好的溶解性,而且稳定性良好,取得了满意的结果。
对于一个方法的可靠性,我们往往必须作方法学上的验证,方法学上的验证方法学包括:线性、精密度、最小检测限、系统适用性.一般来说都是从一个比较适中的浓度做起,然后以10倍量来进行稀释,拟合的时候,一般以相邻的三个点来进行逐级拟合,这样就可以比较方便的确定线性范围,本实验采用的逐级稀释法,从实验数据来看,我们所得到的数据是可靠的,所测定的结果在线性范围内,精密度在允许的范围内,最小限,系统适应性均符合标准。所以,我们所建立的甲醇,乙酸,DMF残留量的GC检查方法,在方法学上准确可靠的,可用于RS中甲醇,乙酸,DMF残留量的检查。
由于我们采用的是外标法,对于气相的定量分析来说,要求峰性能要好,进样的准确性要求特别严格,虽然不一定每一个峰都必须走出来,但是如果操作不当,将导致目标峰出现肩峰,拖尾峰,前沿峰等,这会影响最终的定量结果,所以在实验中准确的操作是非常重要的。
参考文献
[1] 国家药典委员会,中华人民共和国药典20xx年版二部附录[S].北京:化学工业出版社:20
[2] LI HUI-yi(李惠义),WEI jing-jing(魏京京),ZHU Wei-hua(朱维华).GC Detection of Residual Solvents in Iguratimod(艾垃谟德中残留溶剂的检测方法的研究).Chin J Pharm Anal,2004;24(4):422
[3] 林华庆,张蜀,吴素洁等. 毛细管GC测定石杉碱甲贴片中有机溶剂残留量. 2005;9(1):42
[4] 骆传环,黄荣清,邢成等. 沙纳唑含量测定及体外放射增敏作用. 中华放射医学与防护杂. 2005;95(2):57
[5] 赵喆. 毛细管气相色谱法测定辛伐他汀中5种有机溶剂残留量. 药物分析杂志,
12
2006;26:34
[6] Bull RJ,Health. Hazard associate with organic chemical in drinking water Application of short-term biassays in the analysis ofcomplexenviromental mixture, 1981;(2)
[7] 柳庸行.气相色谱在环境监测中的应用.北京:中国环境科学出版社,1998;27(7):45
致 谢
感谢军事医学科学院黄荣清研究员对我不倦的教诲。渊博的知识、敏锐的科研思路以及对前沿科研领域的宏观把握,让我看到作为一个真正研究者身上所特有的品质,他不仅在学业上给予我授业、解惑,在做人的道理上更是对我不断的言传身教,让我更加明白努力乃是成功的必由之路。在此向尊敬的导师表示我最诚挚的谢意。
感谢肖炳坤助理研究员和梁晓东博士的悉心指导和无私关怀。两位老师全面的知识,严谨的的治学风格、和对科学事业孜孜不倦的探索,是我一生都应该学习的。在实习期间,他们给予我极大的帮助,让我少走弯路,少犯错误。对于即将走向职业道路的我来说,这是多么的重要。在此对辛勤耕耘的老师致以崇高的敬意和衷心的感谢。
感谢杨建云书记对我关心和照顾,杨老师为人诚恳,热心助人,在生活工作中对我都非常关照,细心的杨老师总是教导我们要耐心的对待我们生活工作中遇到的问题。要从小事做起,见微知著,让我受益匪浅。在此向杨老师鞠躬来表达我的尊敬和谢意。
感谢军事医学科学院研究生刘力、王晶、胡增峰,在实际工作中,我跟刘力师兄的课题,师兄们耐心的帮助我这个新手,可我却没能帮上什么忙,反而添了不少麻烦,而你们总是对我如此宽容,在此对你们表示我发自内心的感谢。
感谢军事医学科学院二所七室李幸姝、秦芳芳、杨文才和曹玉秀等同学同志的热情帮助和支持。
感谢学院药学系领导和老师们对我的关心和帮助。
感谢家人对我学业的支持!
13
文献综述
CGC及其在有机溶剂残留测定中的应用 摘要:本文综述了气相色谱技术的原理,色谱条件的选择,以及在有机溶剂残留量测定中的应用。
关键词:GC;有机溶剂残留测定
1 概述
毛细管色谱法是50年代开始提出,70年代自从玻璃毛细管和石英毛细管的发展,毛细管色谱法得到了突飞猛进的发展[1],它是通过在毛细管内填充,柱壁涂布,键合或交连用固定相。用气体作流动相的一种分析模式。它具有分离效能高,柱渗透性好,柱容量小,易实现气相色谱-质谱联用和应用范围广的特点,特别在医药领域中,有机溶剂残留的测定有很好的应用。
药物的使用安全是一件跟我们关系非常密切的事情。在大多数化药的合成过程中,不可避免的会使用到一些有机溶剂,如甲醇,乙酸等等。这些有机溶剂不仅影响药物的贮存,药效的正常发挥,更重要的是可能危及用药者的人身安全,所以控制好这些有机溶剂的含量是非常必要的,国家药物质量标准中就有对药物有机溶剂残留量的检测标准
[2]。
由于在药物中含有的有机溶剂可能很多,而且它们的含量往往比较低,所以在对它的检测时要求灵敏度比较高。对数据处理系统的处理能力也是有比较高的要求的。现代GC色谱技术随着仪器的不断完善与发展,虽然在70年代以后,其理论已经基本成熟。但检测技术的成熟与推广,检测器种类的多样性,使其应用范围越来越广。由于GC检测灵敏度高,分离效能好,使之在药物分析领域的应用越来越受到重视[3]。近几年来,除了在传统的挥发油、脂肪油等方面的分析测定不断发展与普及外,在有效成份的研究、药物的比较鉴定、资源的利用,尤其是与药效研究相结合的研究及药代动力学研究方面也得到了开展,在有机溶剂残留测定方面GC有其不可替代的优势[4]。GC与MS的联用技术的推广应用更是把这两者的优势有机的结合在了一起。把分离技术与结构鉴定结合成为药物分析非常重要的手段。在药物分析领域发挥着越来越重要的作用[5]。 2 原理
毛细管色谱法的原理与普通的色谱法理论是基本相同的,但是由于毛细管本身的一
14
些特点,使其在某些方面有些不同。
19xx年Glory提出了毛细管柱的速率 理论方程式,是在Van Deemter 方程式的基础上改进而来的,称为Glory方程式:
H=B/u+Cgu+C1u[20]
式中各项的物理意义及影响因素与填充柱的速率方程式相同,但是由于毛细管是空心的,故其速率理论方程式中涡流扩散项为零;纵向扩散中的弯曲因子为1,传质阻抗项与填充柱的速率方程相同。其精确表达式为
H=2Dg/u+ur2(1+6k+11k2) /24Dg(1+k)2+u2kD2f/3(1+k)2D1[6]
从中看出,纵向扩散项随着载气线速度的增加而很快下降,这是因为线速度大,溶质扩散的时间短。随着载气线速度增加,传质阻抗项增加。
通常总柱效可达到104-106塔板数[7]
3 装置
毛细管气相色谱法与一般色谱法的不同就在于其色谱柱不一样,其他的和填充柱时没有差别的,主要有进样系统,载气,色谱柱,检测器。进样有手动进样和顶空进样,顶空进样准确而且对系统污染很小。手动进样对操作者的要求高,稍有迟疑,都有可能导致峰的延展。[8]目前的检测器有氢离子化检测器FID,热导检测器TCD,电子捕获检测器ECD,火焰光度检测器FPD,热离子化检测器TID,根据所要测定物质的性质来进行选择。
[19]
4 影响因素和条件的选择
从Glory方程式可以看出,H与内经平方成正比,即内经越细,柱效越高。同时也可以看出,Uopt与柱内经成反比,即内经越小Uopt越大。所以就快速分析而言,我们多采用细内经,短毛细管柱。但是内经的变小在实际应用中会受到仪器,操作条件的限制 :
(1)柱内经细导致柱容量小,这使得仪器的灵敏度降低。(2)整个系统的流失、污染和鬼峰应尽量排除。(3)检测器的死体积很小,要加尾吹气。(4)样品容量低,需要特殊的进样装置。所以我们在选择色谱柱时应该多方面的考虑。选择实用的条件,而不是越细越好[9]。
载气在气相色谱条件中飞长重要,它直接关系到仪器地灵敏度,检测限的参数。在毛细管色谱中一般以下列3个因素作为参考依据:(1)分离效能和分离速度;(2)检测器的适应性和灵敏度;(3)载气的物理化学性质,如纯度,安全和价格等[10]。
在实验中,现在多采用气体发生器来制备气体,这又它的优点,同时也存在一些不
15
足,使用气体发生器提供气体,通常比较方便、安全、价格也便宜,但是通常他们的纯度不及瓶装气体,当然不同的发生器的会有一些差异[11]。在我们进行选择时,如果我们对实验的结果要求很高,而且要求灵敏度高,不出现鬼峰的话,通常我们可以采用瓶装的高纯气体。载气速率的选择可以通过Glory方程式计算得到。目前,可选择的载气主要有H2和N2,H2 和N2在最佳柱效上相同。但是,氢气的最佳流速比氮气大四倍,所以近来,多采用氢气作载气[12]。
毛细管色谱有多种固定相可以选择,基本上从物质的极性出发。键合膜的厚度在实验中有很大的影响,膜增厚,导致分离效能降低等。所以,柱直径和膜厚度需要与柱容量和柱效综合考虑[13]。
5 在有机溶剂残留测定中的应用
毛细管气相色谱法的诸多优点,它比较符合药品中有机溶剂残留量的测定要求:痕量,快速,简便。越来越多的固定相选择让应用更为广泛[14]。
在有机溶剂残留量的测定中,要进行的工作的工作主要有,首先选择合适的溶剂对样品进行溶解,通常我们用水来做溶剂,因为它对最后的检测结果使没有影响的,如果水对待测定物质的溶解性不好,那么可以选择其他溶剂,比如DMF,DMSO等非质子性溶剂来溶解[15]。同时考察精密度,系统适应性测试,最低检测限,最低定量限等作为方法学确证的依据,在实验中没有方法学的验证将导致其他人无法重复其实验的可行性。[16]
6 小结与展望
GC技术已越来越趋于成熟,它的高柱效、高分离性能随着计算机技术的飞速发展亦越来越强大与精确,GC与MS的联用技术是今后发展的方向[17],它在对可挥发性未知成分与微量成分的定量及定性检测有其独到之处。除上述应用外,还应用于氨基酸及农药残留量等的分析。随着国家对药品质量越来越规范的管理与要求,GC/MS联用技术在中药指纹图谱的研究上也将发挥其应有的作用,对今后药物研究及药物质量控制的规范化与现代化发展有着重要意义。[18]
参考文献:
[1 ] 郭长强,苏德民,程立方等. 蔓荆子不同炮制品挥发油GC - MS分析[J] . 中草药, 1996;27(9):521
[2] 李洪玉,孙静芸. 气质联用法对没药炮制前后挥发油主成分的比较研究[J]. 中成
16
药,1998;20(9):19
[3] 卢兖伟,王 琦,李胜群,等. 气质联用法分析炮制对乳香挥发油的影响[J] . 中成药, 1996;18(11) :20
[4] 林励,徐鸿华,陈洁标,等.不同引种地大风子仁质量研究. 中国中药杂志[J] .1996 ,21
(2):81
[5] 钟凤林,杨连菊,吉力,等. 不同产地和品种川芎中挥发油成分的研究[J] . 中国中药杂志,1996;21(3) :147
[6] 罗辉,蔡春,张建和,等. 不同产地高良姜挥发油化学成分的比较[J]. 时珍国药研究, 1998;9(1) :29
[7] C. Grote, J. Pawliszyn, Anal. Chem. 69 (1997) 587–598.
[8] C. Camarasu, M. Mezei, A. Szabo, Acta Pharm. Hung.
[9] 郑筱萸. 化学药品和治疗用生物制品研究指导原则[M]. 北京:中国医药科技出版社, 2002.51
[10] 王永铭,李瑞主编.临床药理学. 上海:上海医科大学出版社,1991.30~32
[11] 屠锡德,张钧寿,朱家壁主编. 药剂学. 第3版.北京:人民卫生出版社,2002.1219~1221
[12] 杨雁莉,范兵,王慧川. HPLC测定克拉霉素及其制剂的含量,华西药学杂志,2003;05
[13] 王金芳,周红,齐萍萍,西北药学杂志,2000;Vol 15(6)
[14] 张正奇主编. 分析化学. 北京:科学出版社,2001
[15] 达世一.色谱连用技术,北京:原子出版社.2001
[16] 孙传春.毛细管色谱法.北京:科学出版社.1998.
[17] 骆传环,内部资料,军事医学地科学院.
[18] 郭怀中,孙毓庆, 毛细管色谱在药物分析中的应用.药物分析杂志,1998;9(3)735-736
[19] 胡平,王义明,毛细管李子交换色谱的分离行为. Chin J Anal Chem, 1997;26;1332
[20] R. Shaw, S.A.-M. Smith, C. Nelson, S. Scypinski, poster presentation, American Association of Pharmaceutical Science Conference, June 1994.
17
译 文
英文原文:
Headspace SPME method development for the analysis of
volatile polar residual solvents by GC-MS
Costin C. Camarasu *
Abstract
A solid-phase microextraction (SPME) method has been developed and optimized for the polar residual solvent determination in pharmaceutical products. Five different
polymer-coated fibers were investigated and the Carboxen: polydimethylsiloxane was found to be the most sensitive for all components. Two Headspace SPME methods were developed and optimized: one for the extraction from aqueous solutions, and the other for the extraction from organic solutions (N,N-dimethyl formamide (DMF) and dimethyl sulfoxide (DMSO). The optimum equilibrationtime for all components and all systems was 30 min. It was found that the sample headspace volume has an important effect on method sensitivity and precision. At low headspace volumes (less than one-third of the vial volume),sensitivity improves but at the same time, precision worsens. For 10 ml headspace vials, the optimum headspace volume was found to be 3 ml. The total volatile organic content in the sample also has an important effect on method sensitivity and precision. At low organic content, sensitivity increases but precision drops significantly. Over 0.5% volatile organic content in the sample, the system becomes unstable due to stationary phase swelling by the organic components, and also the sensitivity of the method is drastically reduced. The optimum range for total volatile organic content was found to be between 0.01 and 0.1%. The added Na2SO4 quantity increases the extraction yield. It was found that slightly pressurizing the headspace vial improves the sensitivity of the method by a factor of 2. For the organic system, it was found that the addition of 100 ml DMSO or DMF to 50 mg drug substance and slightly pressurizing the headspace vial gives good results in terms of sensitivity and reproducibility. The measured detection limits were between 0.4 and 200 ng:ml, and the relative standard deviation data were between 2 and 9%. The Headspace SPME from aqueous solutions was found to be ten times more sensitive than Immersion SPME and Headspace SPME from organic solutions. ? 2000 Elsevier Science B.V. All rights reserved.
Keywords : Extraction methods; Solid-phase microextraction; Residual solvent analysis
18
1. Introduction
The residual solvent determination in drug substances,excipients or drug products is known to be one of the most difficult and demanding analytical tasks in the pharmaceutical industry. Furthermore, the determination of polar residual solvents from pharmaceutical preparations continues to present an analytical challenge mainly because these compounds are quite difficult to remove from water or other polar solvents.
In the past few years, solid-phase microextraction (SPME) has gained acceptance for
solventless extraction of water samples. In SPME, the analytes are extracted into a stationary phase that is attached to a length of fused silica fiber [1]. The fiber is contained into a
microsyringe for protection and ease of sampling. In SPME, an exhaustive extraction does not occur, but an equilibrium is established, as analytes partition between the stationary phase and the aqueous phase, or its headspace phase occurs. By sampling from the headspace above the sample matrix, SPME can extract a wide range of organic compounds from various matrices
[2–12]. The present work describes an approach for Headspace SPME method development for the polar residual solvent analysis from pharmaceutical preparations [13–15]. The most important step for successful residual solvent analysis is the development of a stable, selective, sensitive and precise method of analysis of compounds with different volatility and polarities. The gas chromatography-mass spectrometry (GC-MS) technique provides the required
selectivity. Furthermore, the development of a Headspace SPME method requires careful and extensive optimization of the main experimental parameters involved in the extraction and desorbtion process. Earlier [14], we found that extraction time, headspace volume and total organic content have a critical influence on the extraction yield needing to be extensively optimized every time a SPME method is developed. At the same time, the chromatographic conditions, the injector desorbtion temperature and the injection depth also have significant influence on the reliability of the analytical data but, once optimized, the found optimum parameters can be employed for other SPME analytical methods. We also investigated the residual solvent analysis for drug substances that are not soluble in water, and we developed a Headspace SPME method that uses organic solvents instead of water. The Headspace SPME method was compared with an Immersion SPME method. At the same time, we were
interested in the possibility of employing the Headspace SPME sample preparation method from organic solutions from the point of view of suitability for the residual solvent determination method in pharmaceutical products.
2. Experimental
2. 1. Samples and standards
2. 2. SPME de6ice
19
3. Results and discussion
3. 1. Optimization of the Headspace SPME
3. 2. The Headspace SPME method evaluation
4. Conclusions
It is evident from the presented data that extensive optimization is necessary each time a Headspace SPME method is developed. We found that the extraction time, total volatile content, headspace volume, pressure inside the headspace vial and, for organic systems, the added organic solvent quantity are very important parameters. These parameters need to be reoptimized each time a new component is added to the analytical method. At the same time, we found that chromatographic conditions (low starting temperature of the column, 30°C; narrow bore injector liner, 1 min splitless time), together with the optimum desorbtion parameters (optimum injection depth into the injector of 2.5 cm; optimum desorbtion
temperature for CX:PDMS of 300°C, and for all other fibers of 250°C) [14] are not influenced by the type of the extracted compounds and do not need to be reoptimized. We also found that sensitivity and reproducibility are inversely related parameters. When maximizing sensitivity (using low headspace volumes with low total volatile organic contents), the reproducibility worsens. For routine purposes, larger headspace volumes and higher (around 0.1%) total
volatile contents should be used. At the same time, when routine measurements are done, care should be taken that sample solutions and test solutions have similar total volatile organic contents, in the range 0.01–0.1%. Between the investigated polymeric films, the
Carboxen :polydimethylsiloxane - coated fibershowed by far the best sensitivities for all compounds. The fiber was able to extract compounds with different polarity and volatility from aqueous and organic environments. The Carboxen:polydimethylsiloxane- coated fiber showed very good stability in organic media. Between the investigated sample preparation techniques, Headspace SPME from aqueous sam- ples proved to be more sensitive, and Headspace SPME from organic solutions proved to be more precise. The Immersion SPME technique gave similar sensitivities as Headspace SPME from organic solutions and can be replaced with the later one. At the same time, Headspace SPME from organic solutions gave better peak shapes than from aqueous solutions. The SPME GC-MS proved to be a powerful technique in the identification and determination of unknown solvent residues in
pharmaceutical products. With this technique, we were able to identify residual solvents in our proprietary pharmaceutical products. Even if SPME techniques are not yet accepted as sample preparation methods by Pharmacopoeias, taking into consideration their precision, accuracy and speed of analysis, we can state that they are suitable for qualitative:quantitative residual solvent determination in pharmaceutical products.
20
中文翻译:
顶空SPME气-质联用技术在可挥发极性残留溶剂分析中的进展
(节选)
作者 Costin C .Camarau*
摘要:固相微萃取技术(SPME)已经发展并优化用于对药品中的极性溶剂残留量的测定。已经研究的五种不同聚合涂层纤维中,我们发现羧基/聚合二甲基硅氧烷对所有化合物是最为敏感的。我们建立了两种顶空固相微萃取技术并进行了优化,一种是从水中萃取,另一种是从有机溶剂(N,N-二甲基甲酰胺(DMF)和二甲亚砜(DMSO))中萃取。对于所有的化合物和所有的系统,最佳平衡时间均为30分钟。我们发现样品顶空体积对方法灵敏度和精密度有重要的影响。低顶空体积(小于1/3顶空瓶体积)时,灵敏度提高但精密度降低。对10ml的顶空瓶来说,最佳的顶空体积是3ml,样品中总挥发有机溶剂量对方法的灵敏度和精密度也有重要的影响。低有机溶剂总量时,灵敏度增加而精密度显著降低。样品中的挥发有机容量超过0.5%时,系统会变得不稳定,那是因为固定相吸胀有机化合物造成的。同时方法精密度也会彻底的降低。我们发现,最优的有机挥发溶剂总量应该在0.01-0.1%的范围内。外加硫酸钠可以增加提取率。轻微的增大顶空瓶的压力可以使方法的灵敏度提高2个级数。对于有机系统而言,在药物中额外添加100微升DMSO或50微升DMF,或者轻微增大顶空瓶的压力可以在灵敏度和重现性上获得好的结果。检测限在0.4-200ng/ml,RSD在2%到9%之间。顶空固相微萃取技术在水中比浸入固相微萃取技术或顶空固相微萃取技术在有机溶液中的灵敏度高10倍。
关键词:萃取方法;固相萃取;残留溶剂分析
1 简介
众所周知,在制药工业中,原料物、赋形剂、药品中的残留溶剂分析是一件非常困难和烦琐的工作。而且药品中极性有机溶剂的定量分析更是药物分析中的一个挑战,因为这些有机溶剂不容易从水或其它极性溶剂中除去。
在最近的几年里,固相微萃取技术在水不溶性物萃取中已经被接受。在固相微萃取中,提取的待分析物加入到一定长度以熔硅石纤维为支撑的固定相中。这种纤维放入微量调节注射器中可以保护和缓解顶空瓶的压力。在固相微萃取中,绝对的萃取是做不到
21
的,但是可以建立待分析物在固定相与水相二者之间的平衡,或者在顶空部分进行,通过从母液上方的顶空部分抽取,固相微萃取可以从母液中萃取一系列浓度的化合物。
目前的工作可以描述为发展顶空固相微萃取法以用于药物中残留极性溶剂的分析。成功的有机溶剂分析的关键步骤就是找到一个稳定、有选择性、灵敏和精密的有机溶剂化合物的分析方法。气-质联用技术提供了必要的选择性。而且,顶空固相微萃取法的发展要求对试验参数进行谨慎而广泛的优化,包括萃取和解吸附过程。不久前,我们发现萃取时间、顶空体积和总有机容量对提取率有重要的影响,当一个SPME方法建立时,提取率是需要进行广泛优化的。同时,色谱条件、进样解吸附温度和进样深度也对分析数据的稳定性有显著的影响,但是,这些条件一旦被优化,就可以用于其它SPME分析方法。
我们也考察了水不溶性药物有机溶剂的分析。同样,建立了一个用有机溶剂代替水的SPME方法。
顶空SPME方法与浸入SPME方法类似。同时我们对引入顶空SPME样品方法来测定可适用的药品溶剂残留量感兴趣。
2 试验
2.1 样品与标准物质
2.2 顶空固相微萃取设备
2.3 气-质联用
3 结果与讨论
3.1 顶空固相微萃取法的优化
3.2 顶空固相微萃取法的评估
4 结论
很明显,从目前的数据来看,对顶空固相微萃取法的进一步优化是必要的。我们发现,萃取时间、总挥发物容量、顶空体积、顶空瓶内压、对于有机系统来说,额外的有机溶剂量是非常重要的参数,这些参数在分析系统中加入某个新的组份时需要重新优化。同时,我们发现色谱条件(低起始温度,30°C;薄的进样衬垫,1分钟平衡时间)和最好的解吸附作用(2.5cm的最优吸取样品深度;对CX/PDMS最优解吸温度300°C,和对其它纤维的250°C)参数,这些参数不会受提取物的种类的影响,它们是不必再优化的。我们也发现灵敏度与重现性是反相关参数。当最大灵敏度时(用低顶空体积和低总挥发有机溶剂量),重现性会变差。对于常规目的而言,大顶空体积和高的总挥发溶
22
剂量(大约0.1%)是可以接受的。同时,当用常规的用量时,必须注意样品溶液和待测溶液的总挥发有机量应该相近,在0.01-0.1%范围内。
在对聚合物薄膜的研究中,发现羧基/聚二甲基硅氧烷涂层纤维是目前对所有化合物最敏感的。纤维可以从水和有机环境中提取不同极性和不同挥发度的化合物。在有机媒介中,羧基/聚二甲基硅氧烷涂层纤维表现出非常好的稳定性。
对样品前处理技术的研究中,从水溶性样品中进行顶空固相微萃取法被证明更为灵敏,而从有机溶剂中的顶空固相微萃取法则是更为准确。在有机溶剂中,浸入固相微萃取技术有着与顶空固相微萃取同样的灵敏度,而且可以被后者取代。同时,顶空固相微萃取技术在有机溶剂中要比在水中给出更好的峰形。
在药品的未知有机溶剂残留定性鉴定和定量测定中,固相微萃取气-质联用技术被证明是一个强大的技术。我们可以用它来鉴定所有药品中的残留溶剂。即使固相微萃取技术还没有作为样品制剂方法而被药典所接受,但从它们的精密度、准确度和分析速度等方面来考虑,可以说,它们是适用于药品中溶剂残留的定性/定量检测的。 参考文献
(略)
23
-
药学系本科毕业论文范本
药学系本科毕业论文范本封面:学号:(宋体,小4号)福建中医学院届药学系本科生毕业论文(宋体,4号,居中)中文题目(宋体,小2号,居…
-
药学本科毕业论文格式要求
延边大学成人教育本科生毕业论文(设计)撰写规范为了保证我校成人教育本科生毕业论文(设计)质量,提高其严谨性和规范性,特制订下列规范…
-
药学专业毕业论文格式
浙江大学远程教育学院本科生毕业论文设计题目专业学习中心姓名学号指导教师20年月日药学1论文摘要药品集中招标采购推行5年来在规范医药…
-
药学系本科毕业论文格式要求及标准
药本毕业论文格式要求中文宋体英文TimesNewRoman封面按照论文标准要求目录小三黑体目录内容小四宋体2倍行距论文题目三号宋体…
-
药学毕业论文范文.doc1
药学服务与医用指导【摘要】药学服务是一种实践,不仅仅只停留在理论上,同时必须在患者治疗过程中实施并获得效果,不管是预防性的,治疗性…
-
医学系药学专业毕业论文模板
医学系药学专业09级同学:一、毕业论文课题某制剂制备工艺研究或探讨;某制剂某工艺研究或探讨等二、毕业论文内容和目标通过在制药企业实…
-
药学的毕业论文格式
20xx电大届秋季药学专业毕业论文浅谈药品不良反应与安全用药李永梅【关键词】合理的用药随着社会的发展,如何安全、有效、合理的用药已…
-
药学系本科毕业论文范本
药学系本科毕业论文范本封面:学号:(宋体,小4号)福建中医学院届药学系本科生毕业论文(宋体,4号,居中)中文题目(宋体,小2号,居…
-
药学毕业论文范文.doc1
药学服务与医用指导侯永芹自踏入医学殿堂的那一刻起,我便深刻的认识到,“精医术,懂人文,有理想,能创新”是新时期下的药学工作者所应具…
-
指导药学专业本科毕业论文体会
指导药学专业本科毕业论文体会毕业实习在培养高素质、高质量医药学人才过程中有着不可替代的重要作用,是整个本科阶段理论学习向掌握更高阶…
-
药学毕业论文
论药学服务对社会的作用通辽职业学院药学院08级药学八班某人指导教师:马缰【摘要】药学服务是一种实践,不仅仅只停留在理论上,同时必须…