毕业论文样板_最新
基因表达数据分析的方法
摘要:基因表达数据的一个重要应用是给疾病样本分类,如鉴别白血病的类型。而对成千上万个基因表达进行分析,必产生总量巨大的数据集。近年来,支持向量机(SVM)的理论已经取得重大进展,其算法实现策略以及实际应用也发展迅速,开始成为克服“维数灾难”和“过学习”等传统困难的有力手段。利用这一技术分析与整理这些基因表达数据,已有效地解决了生物信息学上这一海量数据的瓶颈问题。本文就支持向量机在基因表达数据分析方面的算法和应用进行了介绍和分析。
关键词:生物信息学;基因表达数据;支持向量机
Methods of gene expression data analysis
Abstract:Gene expression data has an important application to the classification of disease samples, such as identifying the types of leukemia. The analysis of thousands of gene expression data, will produce a tremendous amount of data sets. In recent years, support vector machine (SVM) theory that significant progress has been made towards its strategy and practical applications of algorithms has been developing rapidly and became overcome the "Dimension disaster" and "Over-study", a powerful means of the traditional difficulties. Using this technology analysis and collation of these gene expression data have been effectively solved bottleneck on the enormous bioinformatics data. This paper discusses the algorithms and application of support vector machine in gene expression data analysis.
Keywords:Bioinformatics ;Gene expression data; Support vector machine
目 录
1 引言 ……………………………………………………………………………(1)
2 生物技术的发展前景 …………………………………………………………(1)
2.1生物信息学的研究现状 …………………………………………………(2)
2.2 基因芯片与基因表达数据 ………………………………………………(2)
2.2.1 基因芯片 ………………………………………………………………(3)
2.2.2 基因表达数据 …………………………………………………………(3)
3 基因表达数据分析的方法 ……………………………………………………(3)
3.1 支持向量机 ………………………………………………………………(4)
3.1.1 支持向量分类 …………………………………………………………(4)
3.1.2 分类问题的识别算法 …………………………………………………(4)
3.1.3 支持向量机模型 ………………………………………………………(6)
3.2 支持向量机在基因表达数据分析中的应用 ……………………………(6)
3.2.1基因的选择:t统计法 ……………………………………………………(7)
3.2.2 降维方法:PCA和PLS …………………………………………………(7)
3.2.3 分类结果和评价 ………………………………………………………(8)
4 结论 ……………………………………………………………………………(12)
致谢………………………………………………………………………………(13)
参考文献 …………………………………………………………………………(13)
基因表达数据分析的方法
数学与应用数学专业本科函授校内班 李冠斌
指导教师:宋杰 副教授
1 引言
随着人类基因组计划的完成,人们逐步关注不同人群、正常与疾病状态下DNA序列的变化。DNA序列的变化是有机体种属之间存在差异或种属内存在差异的根本原因,也是影响有机体正常状态和疾病状态的关键因素,对这些基因型差异进行定位、识别以及分类有着重要的定义,这是研究基因型变化与表型变化关系的第一步,是有针对性地预防和治疗疾病的基础。单核苷酸多态性(SNP)[1]是人类基因组中最常见的一种变化。
获得一个基因的序列之后,下一个问题自然就是:怎样利用已知的基因组序列来认识该基因产品的作用是什么?为了了解一个基因的功能,必须知道该基因在什么时候、什么地方表达,其表达所需要的环境条件是什么?也就是要知道该基因所对应的mRNA产生的时间和环境条件以及mRNA的数量。弄清基因在不同组织中、不同条件下及不同的发展阶段的转录丰度,对于解决上述问题是非常重要的。尽管mRNA不是基因的最终产物,但转录是基因法则的第一步,而且认识基因调节网络需要了解转录水平信息。
通过测定基因在某一器官中,不同条件下、不同的发展阶段和不同的组织中的转录水平,可以建立基因表达谱,用以描绘基因组中每一个基因的动态功能。基因表达矩阵是用来描述基因表达数据的矩阵,行代表基因,列代表样本(如:不同的组织,发展阶段和处理);每个格子的数字表示某一基因在某组织(发展阶段或某种处理)中的表达水平。建立这样的矩阵有助于给疾病样本分类,如鉴别肿瘤的类型,以达到最大疗效同时使毒性最低。
2 生物技术的发展前景
生物技术是20世纪末期,在现代分子生物学等生命科学的基础上,发展起来的一个新兴独立的技术领域,已被广泛应用于医疗保健、农业生产、食品生产、生物加工、资源开发利用、环境保护,对农牧业、制药业及其相关产业的发展有着深刻的影响,成为全球发展最快的高新技术之一。
2.1生物信息学的研究现状
生物信息学(Bioinformatics)是一门新兴的交叉学科。它所研究的材料是生物学的数据,而它进行研究所采用的方法。则是从各种计算技术衍生出来的[2]。
20世纪50年代,DNA双螺旋结构的阐明开创了分子生物学的时代。以生物学和医学为主要研究内容的生命科学研究从此进入了前所未有的高速发展的阶段。分子生物学和遗传学的文献积累到90年代中期约40多万篇,到20##年则增长至约50万篇,即在约5年间,增长了10万篇。与此同时,更为大量的数据已经不再以传统的文献形式发表了;这里,最为典型的是DNA序列的数据。至20##年初,国际数据库中记录的接近一千万条DNA序列的碱基数已超过110亿!事实上,现在这一数目已达500亿!在今天的一个大型的基因组测序中心,每天可进行十万个测序反应,产生出107的序列数据。自1999年6月开始进入大规模测序阶段,在短短的8个月内,测序能力上升了将近8倍。至20##年6月,这些中心在6个星期内的测序量就相当于一个人的基因组。也就是说,每周7天,每天24小时,每秒即可产生1000个碱基的数据!随着各国政府和工业界对此的重视,资金大量投入。欧美各国及日本相继成立了生物信息中心,如美国的国家生物技术信息中心(National Center for Biotechnology Informatics,NCBI)、欧洲生物信息学研究所 (European Bioinformatic Institute,EBI)、日本信息生物学中心(Center for Information Biology,CIB)等。NCBI、EBI和CIB相互合作,共同维护着GenBank、EMBL、DDBJ三大基因序列数据库。它们每天通过计算机网络互相交换数据,使得三个数据库能同时获得最新数据。进而促使测序能力的高速上升,使得DNA序列数据每14个月增长一倍!
与上述生物学数据的海量特征相比,生物学数据的复杂特征更具有挑战性。生物学数据的复杂性一方面固然是源于生物体的结构和功能,以及生命活动过程本身的多样性和复杂性,另一方面则是由生物学研究的“社会学原因”所造成的。即生物学的实验数据,一般是在既无标准词法(semantics)、又无句法(syntax)的条件下生成的。这一情况必然进一步加剧生物学数据的复杂性。
生物学数据在海量和复杂性方面所提出的挑战是严峻的。
2.2 基因芯片与基因表达数据
十分幸运的是,在过去的二十多年里,电子计算机芯片对于数字处理的能力的增长基本符合Moore定律(指数增长)。如今的大型计算机的数据处理能力,已经发展到每秒数千亿次乃至数万亿次计算的水平了。有了这一技术支持条件,基因组研究所产生的海量数据,才能够得以有效地加以管理和运行。
2.2.1 基因芯片
基因芯片(gene chip),又称DNA微阵列(DNA micro array),是由大量DNA或寡核苷酸探针密集排列做形成的探针阵列,其工作的基本原理是通过杂交检测信息。基因芯片把大量已知序列探针集成在同一个基片上,经过标记的若干靶核酸序列通过与芯片特定位置上的探针杂交,便可根据碱基互补匹配的原理确定靶基因的序列,通过处理和分析基因芯片杂交检测图象,可以对生物细胞或组织中大量的基因信息进行分析[3]。因而,基因芯片能够在同一时间内分析大量的基因,实现生物基因信息的大规模检测。
2.2.2 基因表达数据
大部分的基因芯片的研究主要是监控基因表达水平,获得基因表达图谱。基因芯片技术是革命性的基因分析,这使得可以监测表达特定组织的基因和比较不同条件下组织的基因表达的等级成为可能,因而,基因表达的数据集已越来越丰富。
基因芯片实验将产生大量的数据,管理与分析这些数据是生物信息学所面临的一个挑战。数据管理的目的是为了更好地利用和共享数据,而数据分析的目标则是从大量的实验数据中提取隐含的生物学信息。特别是对基因表达数据在大规模数据集上进行分析、归纳,可以深入了解基因的功能,理解遗传网络,提供许多疾病发病机制的信息。
然而,计算与检测能力的提高并没有有效地解决生物学的数据问题。海量的数据通过分析与整理后所产生的有用信息(基因表达数据)量变得更巨大,而最大的挑战则是数据分析。基因芯片的表达监控实验产生大量的数据,在这些数据背后隐藏着丰富的信息,需要通过细致的数据分析揭示这些信息,得到有益的结果。但海量的、复杂的基因表达数据使得这一挑战变得不可能。
概括地讲,我们就需要一个好的数据挖掘方法从大型数据库或数据仓库中提取人们感兴趣的、事先未知的、有用的或潜在有用的信息。
3 基因表达数据分析的方法
就生物信息而言,挖掘生物分子数据库已经过二十多年的历程。以前生物信息学的数据挖掘工作主要集中在序列信息方面,而现在通过分析处理基因表达数据挖掘基因功能信息已成为生物信息学研究的一个重点。
数据挖掘常用的方法有:统计分析、聚类分析、决策树、自组织映射、神经网络、遗传算法等[4]。在基因表达数据分析研究中,有一个基本假设,即基因在何时、何地表达的信息携带了关于基因功能的信息。这样,数据挖掘的重要应用就是按照基因表达图谱的相似性分类组织基因。这里主要介绍支持向量机方法。
3.1 支持向量机
支持向量机(support vector machine)是数据挖掘中的一项新技术,它是由Vapnik及其合作者发明。在20世纪90年代中后期得到了全面发展,现已成为机器学习和数据挖掘领域的标准工具。
支持向量机是机器学习领域若干标准技术的集成者。它集成了最大间隔超平面、Mercer核、凸二次规划、稀疏解和松弛变量等多项技术[5]。在若干挑战性的应用中,获得了目前为止最好的性能,开始成为克服“维数灾难”和“过学习”等传统困难的有力手段。
3.1.1 支持向量分类
支持向量分类的目的是开发有效计算的途径,从而能在高维特征向量空间中学习“好”的分类超平面(优化泛化界),而“有效计算”意味着算法能处理的样本数目在100000数量级上。泛化理性论清楚地说明了如何控制容量,因此通过控制超平面的间隔度量可以抑制拟合,而最优化理论提供了必要的数学技术来找到优化这些度量的超平面。而我们可以把这一类分类问题简化为一个最小化权重向量的范数问题。
3.1.2 分类问题的识别算法
统计学习理论是针对小样本情况研究统计学习规律的理论,其核心思想是通过控制学习机器的容量实现对推广能力的控制。
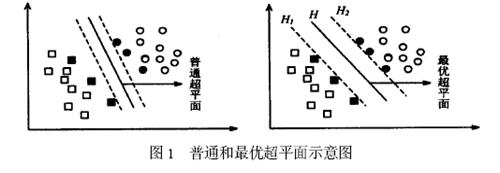
对于训练样本集(x1 , y1), … , (xL , yL) , x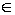 Rn ,y{+1,-1} (L为样本数,n为输入维数),如果训练数据可以无误差地被划分,并且每一类数据距超平面距离最近的向量与超平面之间的距离最大,见图1,则称这个超平面为最优超平面[6]。
Rn ,y{+1,-1} (L为样本数,n为输入维数),如果训练数据可以无误差地被划分,并且每一类数据距超平面距离最近的向量与超平面之间的距离最大,见图1,则称这个超平面为最优超平面[6]。
设最优超平面方程为(w·x)+ b = 0,其中,“·”是向量点积符号。分类判别如下:
yi [(w·x)+ b ] 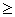 1 ,i = 1,2,…,L (1)
1 ,i = 1,2,…,L (1)
在式(1)中,使等号成立的向量称为支持向量(support vector,SV)。
在2类样本线性可分情况下,求解基于最优超平面的决策数,可以看成解二次型规划问题,即对于给定训练样本,寻找权值w和偏移b的最优值,使得权值代价函数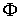 (w)最小:
(w)最小:
min(w)= 0.5‖w‖2 (2)
并满足约束条件(1)。
引入拉格朗日乘子 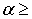 0 ,i = 1,2,…,L 根据Kuhn-Tucker条件,问题可转化为在约束条件(3)下:
0 ,i = 1,2,…,L 根据Kuhn-Tucker条件,问题可转化为在约束条件(3)下:
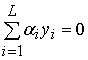 ,
,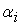 0 ,i = 1,2,…,L (3)
0 ,i = 1,2,…,L (3)
令泛函w(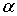 )最大:
)最大:
w()=  -
-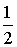
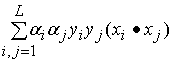 (4)
(4)
设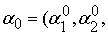 …,
…,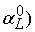 为这个二次型优化问题的解,
为这个二次型优化问题的解,
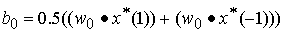 (5)
(5)
式中: 为属于第1类的某个支持向量,
为属于第1类的某个支持向量,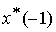 为属于第2类的某个支持向量,则基于最优超平面的分类规划即为指示函数f(x):
为属于第2类的某个支持向量,则基于最优超平面的分类规划即为指示函数f(x):
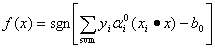 (6)
(6)
3.1.3 支持向量机模型
支持向量机的实现基于如下思想[7]:通过某种事先选择的非线性影射,将输入向量x映射到一个高维特征空间Z,在这个高维空间中构造最优分类超平面,其过程见图2。
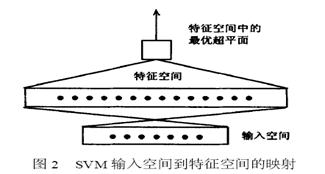
特征空间的维数可能会很高,例如要在一个200维空间中构造一个4或5阶多项式,需要构造一个超过10亿维的特征空间。支持向量机采用内积回旋技术较好地解决这一“维数灾难”问题。
在Hilbert空间中,内积回旋是指: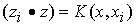 ,其中,Z是输入空间中的向量x在特征空间中的象。根据Hilbert-Schmidt理论,K(x,x
,其中,Z是输入空间中的向量x在特征空间中的象。根据Hilbert-Schmidt理论,K(x,x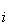 )可以是满足一定条件的任意对称函数。为了在特征空间Z中构造最优分类超平面,并不需要以显示形式来考虑特征空间,只需在输入空间中用非线性决策函数:
)可以是满足一定条件的任意对称函数。为了在特征空间Z中构造最优分类超平面,并不需要以显示形式来考虑特征空间,只需在输入空间中用非线性决策函数:
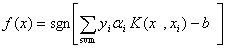 (7)
(7)
它等价于在高维特征空间中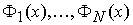 的线性决策函数(K是这个特征空间中内积的一种回旋)。在SVM中构造决策函数式(7)的复杂程度取决于支持向量的数目,而不是特征空间的维数。
的线性决策函数(K是这个特征空间中内积的一种回旋)。在SVM中构造决策函数式(7)的复杂程度取决于支持向量的数目,而不是特征空间的维数。
支持向量机的基本思想可以概括为:首先通过非线性变换将输入空间变化到一个高维空间,然后在这个新空间中求取最优线性分类面,而这种非线性变换是通过定义适当的内积函数实现的。
3.2 支持向量机在基因表达数据分析中的应用
目前,关于表达数据分析方法的研究仍处于起步阶段。但随着技术的成熟和试验控制标准的引入,学界中已出现了多种有针对性的SVM软件,如Proximal SVM(PSVM)对那些预期协同控制的功能分类能够提供较为准确的预测,并能在训练集(Leave-out-one 交叉验证)分类中达到100%的准确率,而在测验集中也能达到不低于79.63%的准确率。但本文的重点不在于软件的应用,而在于数学算法在其中的渗透,因此本文只讨论应用方法。
为了能取得好的分类预报结果,在使用分类方法前使用降维方法,把原来的P维空间降至K维空间,并且满足K<N。然后用支持向量机(SVM)给对降维后得出的主要成分进行分类预报。
3.2.1基因的选择:t统计法
由于基因表达数据通常是测定几十个样本中成千上万个基因的表达值,得到的数据矩阵通常是变量数(基因数)为7000~ 8000,而样本数最多也只能有70~80个。所以直接对这么庞大的矩阵使用降维方法(PCA或PLS)来处理不仅计算量大,而且效果也并不理想。所以,我们首先使用t统计来对原始基因进行筛选。对于一个两类的问题,t统计法的表达式如(8)所示[8]:
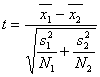 (8)
(8)
其中,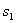 和
和 分别表示两类,
分别表示两类, 为类的均值,N为类的大小,
为类的均值,N为类的大小, 为类的方差。然后,对每个基因计算t值,按t值大小顺序排列。最后取出
为类的方差。然后,对每个基因计算t值,按t值大小顺序排列。最后取出 基因,其中
基因,其中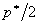 个基因是t值排在最前面的(对应此基因在类1中有高表达值),而另外个基因是t 值排在最后面的(对应此基因在类2中有高表达值)。
个基因是t值排在最前面的(对应此基因在类1中有高表达值),而另外个基因是t 值排在最后面的(对应此基因在类2中有高表达值)。
3.2.2 降维方法:PCA和PLS
主成分分析的中心目的是将数据降维,以排除众多信息共存中相互重叠的信息,是将原变量进行转换,使少数几个新变量是原变量的线性组合,同时,这些变量要尽可能多地表征原变量数据结构特征而不丢失信息。新变量互不相关,即正交[9]。在数学上是通过求x的协方差矩阵的特征向量和特征值来找到低维空间的各个方向,如式(9)所示:
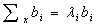 (9)
(9)
式中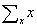 为协方差矩阵,
为协方差矩阵,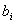 为特征向量,
为特征向量,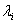 为特征向量所对应的特征值。
为特征向量所对应的特征值。
偏最小二乘与主成分分析很相似,其主要差别在于用于描述变量Y中因子的同时也用于描述变量X。为了实现这一点,在数学上是以矩阵Y的列去计算矩阵X的因子,与此同时,矩阵Y的因子则由矩阵X的列去预测。其数学模型为式(10)和式(11):
X = TP + E (10)
Y = UQ + F (11)
其中,T和U的矩阵元分别为X和Y的得分,而P和Q的矩阵元分别为X和Y的载荷,E和F分别为运用偏最小二乘模型去拟合X和Y所引进的误差。
在理想的情况下,X中的误差的来源与Y中的误差来源完全相同,即影响X与Y的因素相同。但实际上,X中的误差与Y中的误差并不相同。因而, ,但当两个矩阵同时用于确定因子时,则X和Y的因子具有如下式(12)的关系:
,但当两个矩阵同时用于确定因子时,则X和Y的因子具有如下式(12)的关系:
u = bt + e (12)
式中b所表征的即u和t间的内在关系。
3.2.3 分类结果和评价
本预测实验使用re-randomozation方法来评价分类结果和方法的稳定性,这种方法具体有以下步骤:
(1)随机从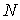 个样本数据集中抽取
个样本数据集中抽取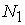 个样本数据作为训练集,这个样本将用来建模,剩余的
个样本数据作为训练集,这个样本将用来建模,剩余的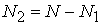 个样本将作为测试集用来验证模型。
个样本将作为测试集用来验证模型。
(2)使用训练集建模,并使用Leave-out-one对模型进行交叉验证。
(3)使用剩余样本对模型进行验证,这可以有效地防止过拟合现象。
(4)为了进一步防止过拟合现象,重复1-3步来验证模型。
这里先引用同济大学俞振超副教授等在这应用方面的一个数据。其使用的数据集是Golub等在1999年发表的急性白血病(Acute Leukemia)数据集[10]。原始的训练集包括38个骨髓样本,其中27个样本是急性淋巴细胞白血病(Acute Lymphoblastic Leukemia,ALL)和11个是急性骨髓性白血病(Acute Myeloid Leukemia,AML)样本,这些样本均来自成人。测试集包括24个骨髓样本和10个血液样本,它们取自成人和孩子(20个ALL和14个AML)。Golub等分别对每个样本测试了6871个基因的表达值。而且,训练集是一个6871行38列的数据矩阵,测试集是6871行34列的数据矩阵。
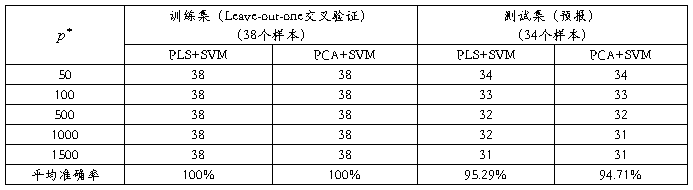
首先使用Golub等使用的方法,得到一个没有负值的数据矩阵。然后,对数据取对数,中心化和归一化。在数据预处理后,我们对训练集选择基因分别取=50,100,500,1000和1500。然后使用PCA和PLS降维,再用SVM分别对各种情况进行数据处理,其中PCA和PLS均取15个主成分,SVM权重均取1:3。对训练集使用Leave-out-one交叉验证,对测试集进行预报,结果如表1所示:
表1 急性白血病数据分类结果
最后我们利用re-randomozation方法检验模型和分类结果的稳定性,分别取和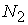 均为36,随机做了100次,结果如表2所示:
均为36,随机做了100次,结果如表2所示:
表2 重复随机学习的急性白血病数据分类结果

从表2我们可以看到,无论是对训练集的交叉验证,还是对测试集的预报,PLS均优于PCA,但PCA的总体预报效果也不错,两者在处理急性白血病数据时效果相差不大。这也是因为我们在选择基因时选择了t统计值最大和最小的,所以被选择的基因通常在类1中表达高时,在类2中必定表达比较低,反之亦然。
然而降维方法(PLS和PCA)和SVM均有固定的标准算法和选值,为了测试此应用的通用性,我选择了另一白血病(Leukemia)数据集(此癌症基因表达分类数据由宋杰副教授提供)。该数据集包含72 个骨髓样本,其中包括47例淋巴性白血病(ALL)样本和25 例急性骨髓性白血病(AML)样本,基因数是7129。此数据包含A和d两矩阵(A是数据,d是类别),其中A是一个72行7129列的数据矩阵,d是一个72行1列的数据矩阵,两矩阵均为双字节的Matlab数据文件。数据处理后,在选择个基因时不再提取t值的最大和最小值,而是选取比较接近这些值的部分即最大和最小部分中随机选取。由此,我们仍然对训练集选择基因分别取=50,100,500,1000和1500,其他训练环境均不改变。我们先从72个样本中随机选取=38作为训练集,剩下的=34作为测试集。对测试集进行预报,结果如表3所示:
表3 白血病数据分类结果
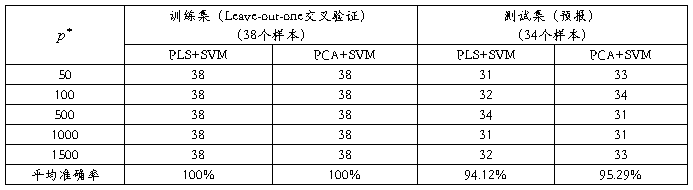
由表3我们看到预报结果仍然理想,而且PCA的预报效果有所提高,并优于PLS。同样利用re-randomozation方法检验模型和分类结果的稳定性,分别取和均为36,再随机做了100次,结果如表4所示:
表4 重复随机学习的白血病数据分类结果
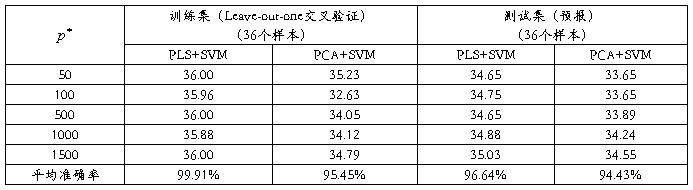
由表4我们可以看到PLS依然优于PCA,但PCA的预报效果有所提升。尽管我们改变了预测的对象,而且对象的构成和规格大不相同,但很幸运地,我们从表3和表4可以发现,在改变了基因表达数据集后,预报结果的准确率基本没有发生太大的变化,甚至在最大和最小部分内改变了所选择的t统计值,准确率依然没有发生质的改变。这大大地证明了此方法的通用性和广泛性。说明了这一分类方法基本适合各种基因表达分类数据,并且在预报过程中对所选基数的精度依赖性不强(即在最优范围内的取值都能作出好的预报效果),使得这一应用方法能够广泛地被使用。因为不需要依赖高精度的计算和严密的测试,从而易于被各类型人员所接受和使用。当然科学是严紧的,基因表达数据的分析必须遵守其标准和规律。
在上面的两个预报中,我们都选择了t统计值的最大和最小部分,这样PLS和PCA在处理的数据本身已有很强的类分辨能力,这相当于在使用PCA降维时也用到了已知的分类信息,因此,使用PLS和PCA对最终的分类效果差别不大。为了检验出PLS和PCA在效果上的差别和t统计值对分类效果的影响,我们在选择个基因时不再取t值最大和最小部分的基因,而是从中间向两边取,例如,当=50时,首先找到t值排在最中间的基因,然后向前后分别取25个。为了更好的对比预报结果,我们依然取同济大学俞振超副教授等应用的数据作为预报结果(即预报对象为Golub等在1999年发表的急性白血病数据集)。我们对38个样本组成的训练集分别取=50,100,500,1000,1500。交叉验证和预报结果如表5所示:
表5 改变选择基因标准后急性白血病数据分类结果
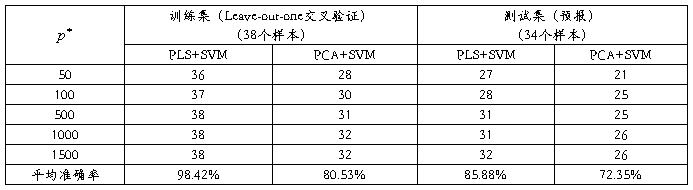
从表5中可以清楚的看到,由于基因选择使用了新的标准使得PLS和PCA处理数据本身对类的分辨能力很弱,也就是数据本身并没有反映样本的分类信息,所以使用PCA对数据处理时它只是找到了一些数据本身方差很大的方向,它与分类信息关系不大,因而在做交叉验证时结果不是很好,也就是显而易见的;虽然PLS处理的数据本身对类的分辨能力很弱,但PLS在处理数据时用到了样本的分类信息,因而,用PLS找到的方法是考虑了样本分类的信息方向,这一点在训练交叉验证中可以看到。用SVM对PLS建立的模型明显优于对PCA建立的模型,从而在预报测试集样本时也明显优于PCA。表5的结果也明确的展现了t统计法在其中的地位。
为了验证模型和结果的稳定性,我们依旧利用re-randomozation方法来验证,分别取和均为36,再随机做了100次,结果如表6所示:
表6 改变选择基因标准后重复随机学习的急性白血病数据分类结果
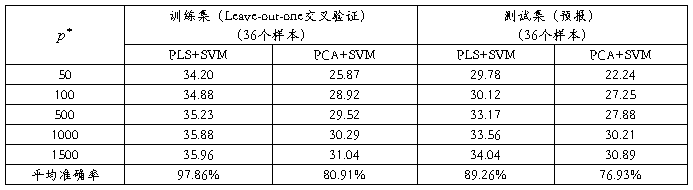
从表6的结果中可以看到模型和结果比较稳定,而且同样反映出PLS明显优于PCA。
4 结论
由于基因表达数据具有变量数(基因数)远大于样本数的特点,本文选择了用t统计法选择基因,并使用降维方法处理数据,最后用SVM进行建模预报。本文就支持向量机在基因表达数据分析方面的算法和应用进行了介绍和分析,并突出了t统计法在当中的地位和比较了PLS和PCA两种降维方法的效果,在文章里给出的是PLS的效果优于PCA,但在t统计法的最优取值范围内两者的处理效果均为较好。
目前,关于表达数据分析方法的研究仍处于起步阶段。例如,目前的相似性检测方法多适用于特定的情况下,广泛适用的相似检验方法仍待系统地研究开发。随着技术的更加成熟和试验控制标准的引入,以及广泛接受的数据标准化和质量控制方法的形成,系统地描绘基因在不同的器官、组织、发展阶段和试验条件下的表达谱是可能的。描绘各种化合物的表达谱,将有助于发现它们可能存在的毒性机制和细胞过程,会产生各种不同的标记。这个过程类似系统基因组测序。可靠地搜索相似的表达谱或发现相关表达谱或发现相关表达谱的共同标记依赖于可靠的算法,所以数学在其中的渗透和应用还有很大的空间,要达到算法的完善,这还有很长的路要走。
致 谢
本文是在导师宋杰副教授的悉心指导下完成的。宋老师高尚的品格、渊博的知识、严谨的作风使我受益匪浅,在此谨向宋老师致以由衷的敬意、真诚的感谢和美好的祝愿。
衷心感谢每一位教导过我的老师,是他们使我拥有良好的专业基础,因而有能力完成这一毕业论文。
衷心感谢我身边的每一位好友。
最后我要感谢我的父母和亲人的支持和鼓励。
参考文献
[1] 赵国屏等. 生物信息学[M]. 北京: 科学出版社, 2002.4.
[2] 陈润生.生物信息学[J]. 生物物理学报,1999,15(1): 6-12.
[3] Ramsay R. DNA chips: State-of-the-art[J]. Nature Biotechnology, 1998.16:40-44.
[4] Michael PSB et al. Knowledge-based analysis of micro array gene expression date by using support vector machine[J]. PNAS, 2000.97:262-267.
[5] (英)克里斯特安尼(Cristianini N)等,支持向量机导论[M]. 李国正,王猛,曾华军译. 北京: 电子工业出版社, 2004.3.
[6] 史忠植. 知识发现[M]. 北京:清华大学出版社,2002.
[7] Vladimir NV. 统计学习理论的本质[M]. 张学工译. 北京:清华大学出版社,2000.
[8] Nguyen DV,Rocke DM. Tumor classification by partial least squares using microarray gene expression date[J]. Bioinformatics, 2002.18:39-50.
[9] 许禄. 化学计量学方法[M]. 北京:科学出版社, 1995.
[10]Golub TR,Slonim D,Tamayo P,Huard C,Gaasenbeed M,Mesirov J,Coller H,Loh M,Downing J,Caligiuri MA,Bloomfield CD and Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring[J].Science, 1999.286: 531-537.
-
毕业论文总结 毕业致词 精选8篇
范文一:毕业论文的写作锻炼了我收集和整合信息的能力,从着手准备论题到实地调查再到整个论文的完成,凝聚着无数人对我的关怀和帮助,没有…
-
汉语言文学专业毕业论文、调查报告写作要求
汉语言文学专业毕业论文撰写要求详细说明一、毕业论文的总体要求1、毕业论文的写作是“中央广播电视大学人才培养模式改革与开放教育试点”…
-
毕业论文期中总结报告
期中总结报告人往往在消费时间的时候感觉不到时间的流逝,而总是等到回忆或总结时才发觉到了时间的脚步。既然如此,人就该经常性的反省自己…
-
20xx级本科毕业论文(设计)工作总结
20xx级本科毕业论文(设计)工作总结毕业论文(设计)是实践教学的重要环节,是本科生综合运用所学基础理论、专业知识和基本技能、接受…
-
法律系毕业论文
辽宁商贸职业学院毕业论文浅析公民的政治权利年级:20xx级专业:法律事务学生姓名:于航学号:20xx03020xx3指导教师:赫荣…
-
生物毕业论文范文
摘要:农业生态系统是以种植业为主题以农田生物群落与无机环境相组合,按照人们经济发展的莆求所构成的人工碱半人工生态系统。农田生物群落…
-
毕业论文范文
湖北生物科技职业学院学生毕业论文论文题目武汉蔬菜生产前景系别园艺系专业园艺技术年级0601姓名胡振成学号0603020xx4指导教…
-
生物技术专业暑期社会实践报告范文
20xx年生物技术专业暑期社会实践报告范文实习目的1通过深入各实习岗位进行综合性的实习将所学的基础理论基础知识和基本技能运用于实践…
-
毕业论文开题报告范文
武汉大学本科毕业论文设计开题报告题目乳液聚合法制备Fe3O4StSMS磁性高分子微球姓名陈超学号20xx2230435220xx专…
-
优秀毕业论文排版范文
农村小学科学概念教学有效性研究以沁水县张马小学为例教育与心理科学系小学教育专业0903班张超指导教师杜艳芳摘要小学科学概念是小学科…
-
重庆亲子鉴定指南之十六哪些样本可以用来进行DNA亲子鉴定
重庆亲子鉴定指南之十六哪些样本可以用来进行DNA亲子鉴定理论上讲所有人体有核细胞都可以用来进行DNA亲子鉴定其中最常用的样本是血液…