药品不良反应事件报告表
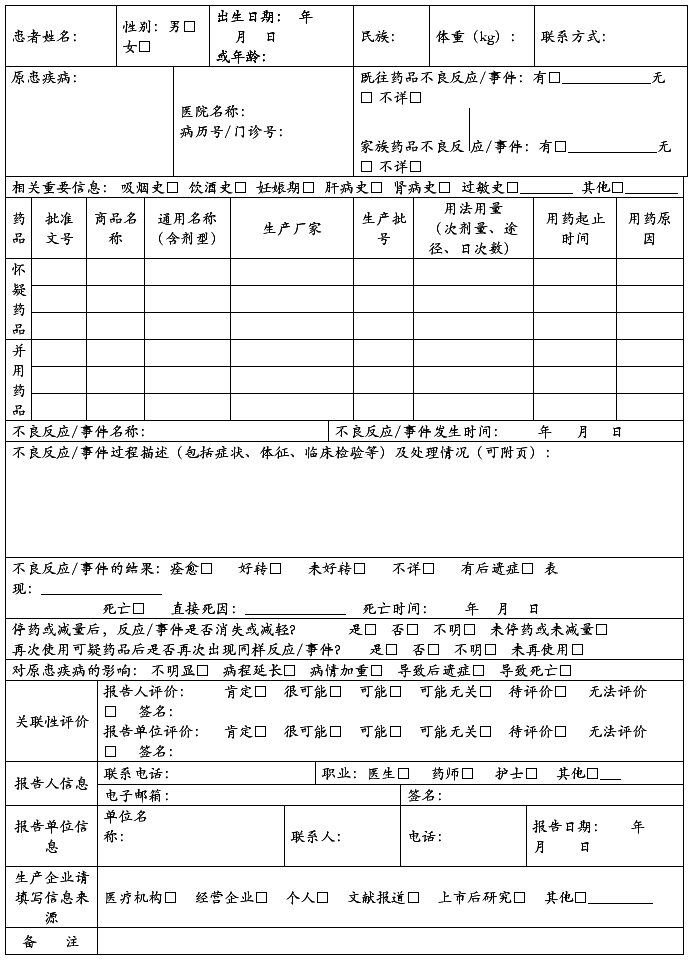
药 品 不 良 反 应 / 事 件 报 告 表
首次报告□ 跟踪报告□ 编码:
报告类型:新的□ 严重□ 一般□ 报告单位类别:医疗机构□ 经营企业□ 生产企业□ 个人□ 其他□
严重药品不良反应,是指因使用药品引起以下损害情形之一的反应:
1) 导致死亡;
2)危及生命;
3)致癌、致畸、致出生缺陷;
4)导致显著的或者永久的人体伤残或者器官功能的损伤;
5)导致住院或者住院时间延长;
6)导致其他重要医学事件,如不进行治疗可能出现上述所列情况的。
新的药品不良反应:是指药品说明书中未载明的不良反应。说明书中已有描述,但不良反应发生的性质、程度、后果或者频率与说明书描述不一致或者更严重的,按照新的药品不良反应处理。
报告时限
新的、严重的药品不良反应应于发现或者获知之日起15日内报告,其中死亡病例须立即报告,其他药品不良反应 30日内报告。有随访信息的,应当及时报告。
其他说明
怀疑药品:是指患者使用的怀疑与不良反应发生有关的药品。
并用药品:指发生此药品不良反应时患者除怀疑药品外的其他用药情况,包括患者自行购买的药品或中草药等。
用法用量:包括每次用药剂量、给药途径、每日给药次数,例如,5mg,口服,每日2次。
报告的处理
所有的报告将会录入数据库,专业人员会分析药品和不良反应/事件之间的关系。根据药品风险的普遍性或者严重程度,决定是否需要采取相关措施,如在药品说明书中加入警示信息,更新药品如何安全使用的信息等。在极少数情况下,当认为药品的风险大于效益时,药品也会撤市。
第二篇:局令第07号附件1药品不良反应/事件报告表
附表1 制表单位:国家食品药品监督管理局
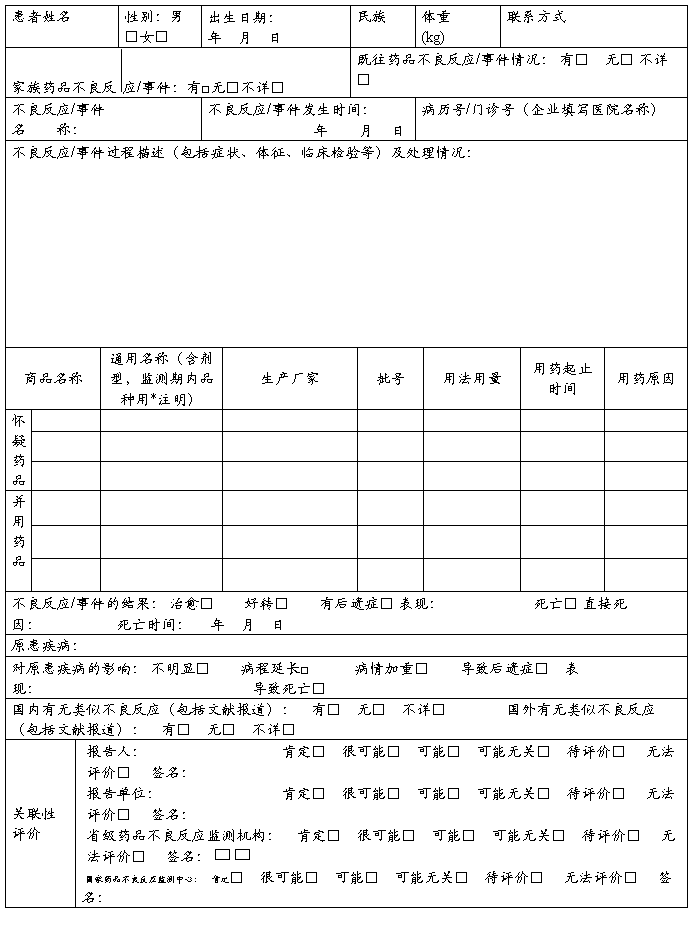
药品不良反应 / 事件报告表
新的□严重□一般□ 医疗卫生机构□ 生产企业经营企业□ 个人□ 编码□□□□□□□□□□□□□□□□□□□
单位名称: 部门: 电话: 报告日期: 年 月 日


报告人职业(医疗机构):医生□ 药师□ 护士□ 其他□ 报告人职务职称(企业): 报告人签名:
◇不良反应/事件分析

◇严重药品不良反应/事件是指有下列情形之一者:
① 引起死亡 □
② 致畸、致癌或出生缺陷 □
③ 对生命有危险并能够导致人体永久的或显著的伤残 □
④ 对器官功能产生永久损伤 □
⑤ 导致住院或住院时间延长 □
◇编码规则:
省(自治区、直辖市) 市(地区) 县(区) 单位 年代 流水号
□□ □□ □□ □□□□ □□□□ □□□□□
注:省(自治区、直辖市)、市(地区)、县(区)编码按中华人民共和国行政区划代码填写。
单位编码第一位如下填写:医疗机构1、军队医院2、计生机构3、生产企业4、经营企业5。
个人报告单位编码一栏填写6000
◇注:通用名称一栏,首次获准进口5年内的进口品种用*注明
国家药品不良反应监测中心 药品不良反应监测中心
通信地址:北京市崇文区法华南里11号楼二层 通信地址:
邮 编:100061 邮 编:
电 话:(010)67164979 电 话:
传 真:(010)67184951 传 真:
E – mail :report@adr.gov.cn E – mail:
新的、严重的药品不良反应/事件病例报告要求
药品生产企业报告要求
1. 填报《药品不良反应/事件报告表》;
2. 产品质量检验报告;
3. 药品说明书(进口药品还须报送国外药品说明书);
4. 产品注册、再注册时间,是否在监测期内(进口药是否为首次获准进口5年内);
5. 产品状态(是否是国家基本药物、国家非处方药、国家医疗保险药品、中药保护品种);
6. 国内上年度的销售量和销售范围;
7. 境外使用情况(包括注册国家、注册时间);
8. 变更情况(药品成分或处方、质量标准、生产工艺、说明书变更情况);
9. 国内外临床安全性研究及有关文献报道情况;
10. 除第1、2项以外,其他项目一年之内如无变更,可以免报。
-
新版药品不良反应事件报告表
药品不良反应事件报告表首次报告跟踪报告编码报告类型新的严重一般报告单位类别医疗机构经营企业生产企业个人其他严重药品不良反应是指因使…
-
药品不良反应-事件报告表
例表1药品不良反应/事件报告表首次报告□跟踪报告□编码:报告类型:新的□严重□一般□报告单位类别:医疗机构□经营企业□生产企业□个…
-
《药品不良反应_事件报告表》及填写说明
药品不良反应事件报告表填写说明一填写注意事项1药品不良反应事件报告是在特定时间的某个具体的患者使用某个批次的药品而出现的药品不良反…
-
药品不良反应事件报告表模板
附药品不良反应事件报告表模板仅为示例完善填写格式和填写事项首次报告跟踪报告编码报告类型新的严重一般报告单位类别医疗机构经营企业生产…
-
药品不良反应报告表
药品不良反应事件报告表报告类型新的严重一般首次报告跟踪报告报告来源医疗机构药品经营企业药品生产企业其他除非得到允许报告表中的个人信…
-
药品不良反应和医疗器械不良事件监测与报告制度
药品不良反应和医疗器械不良事件监测与报告制度一药品不良反应定义上市药品在正常用法用量情况下出现的与用药目的无关的或意外的有害反应包…
- 药品不良反应报告模板
-
药品不良反应报告表
附表1药品不良反应事件报告表首次报告跟踪报告编码报告类型新的严重一般报告单位类别医疗机构经营企业生产企业个人其他13严重药品不良反…
-
药品不良反应事件报告表模板
附药品不良反应事件报告表模板仅为示例完善填写格式和填写事项首次报告跟踪报告编码报告类型新的严重一般报告单位类别医疗机构经营企业生产…
-
药品不良反应报告表填写范例
制表单位紧急一般编号药品不良反应报告表医疗单位使用医院名称解放军第251医院科别呼吸科电话8785120报告日期20xx年1月18…
- 药品不良反应报告范例[1]