原子吸收光谱实验报告
仪 器

分 析 实 验 报 告
学 院: 化学工程学院 专 业: 化学工程与工艺 班 级: 化工091班 姓 名: 学 号 094020101
指 导 教 师: 李颖 日 期: 20xx年5月15日

实 验 名 称: 原子吸收光谱实验
一、 实验目的
1. 了解AA的结构,了解仪器的开、关程序。
2. 了解AA的分析过程
二、 实验原理
原子吸收光谱分析法是基于原子由基态跃迁到激发态时对辐射光吸收的测量。通过选择一定波长的辐射光源,使之满足某一元素的原子由基态跃迁到激发态的能量要求,则辐射后基态的原子数减少,辐射吸收值与基态原子数有关,即由吸收前后辐射光强度的变化可确定待测元素的浓度。
三、 仪器和试剂
仪器: 日本岛津AA-6200 试剂: 蒸馏水、镍标准溶液

四、 实验步骤
以镍溶液标准曲线的绘制及样品的测定为例,实验操作如下:
1. 做好实验前的安全工作。首先打开实验室窗户通风,接着打开总电源启
动排气装置—这里主要考虑到实验所用的乙炔气体的危险性,若在密闭环境下积聚浓度太高就有发生爆炸的可能性。乙炔装在白色钢瓶内。
2. 打开空气压缩机,空气是作为乙炔气体燃烧的助燃气体,它们共同构成
了乙炔—空气燃烧系统。
3. 开气体钢瓶,钢瓶总阀开度不必太大,大概旋转45度角即可,同时气体
的流通还受一个微调阀控制,即总阀开启气体并不一定能通过管路。因此,应同时调节总阀与微调阀,使指示计显示正常稳定的压力值。 这里需说明,微调阀只需在更换气体后的第一次使用调节完成,以后实验只要调节总阀即可。
4. 安装空心阴极灯。空心阴极灯的是根据实验要求来选取的,即测什么元
素就用什么元素的空心阴极灯。空心阴极灯可以从实验室直接拿取,如没有则要提前到市场上购买。AA-6200配备了两个灯座,(HCL-1, HCL-2)这大大提高了实验的方便性,通过灯的轮换装置可以任意切换安装的两盏灯。说明:空心阴极灯的安装应在仪器打开之前完成,因为仪器一旦启动其灯座上可能有电流通过,这时再徒手安装灯就有一定的危险。
5. 预热仪器。将仪器打开后预热半小时,这是保证仪器运转的稳定性,从
而提高测量的精确性。
6. 软件操作。 首先打开软件进行元素选择,可以从下拉式菜单中选取也可
以直接从元素周期表中选择,接着点击”connect” 按钮,这一步主要是仪器进行自检,确定各个部件都能正常使用,然后是进行参数编辑,选择对应的灯号并将空心阴极灯通电,最后搜索波长,波峰的完美与否可能会影响实验的准确性,因此如出现的波形严重偏离对称性,那么可以重复搜索直到可以接受为止。
7. 标准曲线的绘制 。做标准前先点火,同时按住黑白两颗按钮直到火焰完
全燃烧,接着在软件上选择“STD”字样,其个数应取决于用于做标准曲线点的个数,每次做完一个点必须要等仪器显示的进样吸光度为零才
可以做下一个点,如果仪器不能自动归零,此时点击“AUTO ZERO”按
钮即可。 以此方法测完所有的点,标准曲线就完成了。
8. 样品测量。选择“UNK”,其个数与所测样品的数量有关。每次测完一组
样品仪器显示也必须归零才能接着做下一组,以此方法测完所有的样品。
9. 结束实验。首先关闭软件与电脑,接着按住仪器最上面一颗黑色的按钮
3-4秒钟,放出管路中多余的乙炔气后关闭仪器,最后将钢瓶总阀旋紧,同时关闭空气压缩机与总电源。
10. 离开实验室。整理桌面,打扫卫生,关好窗户离开实验室。
五、 实验讨论
1. 本实验以镍溶液为标准绘制标准曲线,同时进行样品的测定,最大吸收
波长为232.35nm。
2. 实验原子化方法有火焰法和非火焰法,本实验采用火焰法。火焰温度越
高越有利于离子的原子化,扩大测定范围,但同时高温产生的热激发态
原子增多对定量不利。在保证测定元素充分还原为基态原子的前提下,
应尽量采用低温火焰,使基态原子的激发依赖于对光的吸收。本实验采
用乙炔-空气火焰,乙炔气体的燃烧,空气作为一种助燃气体使用。因此
存在一定的危险性,实验前必须做好防护措施,比如,开窗通风等。
3. 注意事项:
1) 实验必须严格按照操作顺序进行下去,不可跳级操作或者漏操作,
否则实验难以完成。
2) 实验前应备好蒸馏水,将前次实验留下的蒸馏水进行换洗,否则
在测量时仪器显示的吸光度将难以回到零点,如经常自动回零势
必会增加实验的时间及准确性。
3) 每次实验都必须进行新的标准溶液曲线图的绘制,上次保存的图
本次实验不可用。
4. 关于标准曲线的制作及点的删补法
标准曲线绘制精确以否将直接影响样品测量的准确性,样品离子浓度值通过标准曲线直接由数据处理系统自动获取。在标准曲线的绘制过程中,出现的问题主要是曲线上点的线性度不够好,原则上要求r必须在三个9以上,即r=0.999。
但是实验中往往很难做到这一点,原因如下:一种可能是由于原子分光光度计使用年限过长导致仪器的灵敏度或精确度受到影响;二是由于自配标准溶液没有达到规定的要求,即所配溶液的浓度与实际值有一定的差距致使标准曲线产生误差,这也是主要的原因所在。因此做好这一步必须严格做好标准溶液的配制工作。(原标准溶液的浓度为1000?g/ml,按照实验的要求稀释相应的倍数)。
标准曲线绘制过程中会出现除一两个点外,另外的点线性度都比较好,那么此时就需要补做或者删除。补做的方法是重新配置一份该点浓度的标准溶液再进行标定,因此为节省实验时间,实验前应准备两份同样浓度的标准溶液,在实验中可以择优选取。删除的方法是在对应的浓度值横栏中选择上方的“X”图样,双击两下,该点即可自动在标准曲线图上消失。
5. 实验分析
实验中提示出现问题主要在“Line Search”这步,常显示“气压不足”或者“空心阴极灯能量不足”。气压不足主要是钢瓶中剩余乙炔气体不够无法支持燃烧或者是钢瓶阀门开度不够,但往往是前者原因,因此在每次实验结束后要注意钢瓶中是否有足够的气体来完成下次实验,乙炔气体的更换需要一定的时间,因此事先报告可以加快实验进度。灯能量不足主要是空心阴极灯或是灯座的原因。如果是空心阴极灯由于没有足够预热,那么多做几次即可成功,如果是空心阴极灯由于使用年限过长,那么必须更换新的灯来完成实验。而如果是灯座的原因那只能更换别的灯座,该仪器提供两个灯座,因此可灵活使用。
实验中最典型的操作错误是空心阴极灯的选择与所测元素不符,即测什么元素的浓度必须用该元素的空心阴极灯。空心阴极灯的安装调整必须在仪器打开前完成,(这是因为仪器打开后灯座将有电流通过,不安全)并使用转盘手动完成。 同样必须要记住的是,哪个灯座安装的是哪盏空心阴极灯,如果顺序打乱,那么在检测波长这一步是不能完成的,实验就不能进行下去。
6. 本实验操作时未成功,原因是乙炔气体连接到日本岛津AA-6200的皮管
在实验操作前有脱落过,不能点燃,不能进行原子化,导致实验不能进
行。同时,实验过程中应谨记:实验必须严格按照操作顺序进行下去,不可跳级操作或者漏操作。
7. 原子吸收光谱优点:
1) 选择性强。这是因为原子吸收带宽很窄的缘故。因此,测定比
较快速简便,并有条件实现自动化操作。
2) 灵敏度高。原子吸收光谱分析法是目前最灵敏的方法之一。火
焰原子吸收法的灵敏度是ppm到ppb级,石墨炉原子吸收法绝对灵敏度可达到10-10~10-14克。常规分析中大多数元素均能达到ppm数量级
3) 分析范围广。发射光谱分析和元素的激发能有关,故对发射谱
线处在短波区域的元素难以进行测定。另外,火焰发射光度分析仅能对元素的一部分加以测定。
4) 抗干扰能力强。第三组分的存在,等离子体温度的变动,对原
子发射谱线强度影响比较严重。而原子吸收谱线的强度受温度影响相对说来要小得多。
5) 精密度高。火焰原子吸收法的精密度较好。
8. 缺点:
1) 原则上讲,不能多元素同时分析。测定元素不同,必须更换光
源灯,这是它的不便之处。原子吸收光谱法测定难熔元素的灵敏度还不怎么令人满意。在可以进行测定的七十多个元素中,比较常用的仅三十多个。当采用将试样溶液喷雾到火焰的方法实现原子化时,会产生一些变化因素,因此精密度比分光光度法差。现在还不能测定共振线处于真空紫外区域的元素,如磷、硫等。
2) 标准工作曲线的线性范围窄(一般在一个数量级范围),这给实
际分析工作带来不便。对于某些基体复杂的样品分析,尚存某些干扰问题需要解决。在高背景低含量样品测定任务中,精密度下降。如何进一步提高灵敏度和降低干扰,仍是当前和今后原子吸收光谱分析工作者研究的重要课题。
第二篇:第4章 原子吸收光谱法
第4章 原子吸收光谱法
4.1 内容提要
4.1.1基本概念
原子吸收光谱法──基于待测元素的基态原子在蒸气状态对原子共振辐射的吸收程度来确定物质含量的分析方法。
自然宽度──没有外界影响时,谱线仍有一定的宽度,称为自然宽度。
多普勒(Doppler)变宽──又称热变宽,是由于原子不规则的热运动引起的变宽,通常在原子吸收光谱法测量条件下,多普勒变宽是影响原子吸收光谱线宽度的主要因素之一。
压力变宽──由于原子吸收区气体原子之间相互碰撞导致的谱线变宽。根据碰撞的原子不同又分为:洛伦兹(Lorentz)变宽和赫尔兹马克(Holtsmark)变宽。
洛伦兹变宽──是指待测元素原子与其它种粒子碰撞引起的变宽,它随原子区内气体压力增大和温度升高而增大。
赫尔兹马克变宽──是指待测元素原子之间相互碰撞而引起的变宽,也称共振变宽,只有在待测元素的浓度高时才起作用。
积分吸收──在吸收线轮廓内,吸收系数的积分称为积分吸收系数,简称积分吸收,它表示吸收的全部能量。积分吸收与原子蒸气中吸收辐射的原子数成正比。
峰值吸收──吸收线中心波长处的吸收系数为峰值吸收系数,简称峰值吸收。在温度不太高的火焰条件下,峰值吸收系数与火焰中待测元素的原子浓度成正比。
锐线光源──是发射线半宽度大大小于吸收线半宽度的光源,如空心阴极灯。
化学计量焰──由于燃气与助燃气之比与化学反应计量关系相近,又称其为中性焰。这类火焰温度高、稳定、干扰小、背景低,适合于许多元素的测定。
富燃焰──指燃气大于化学计量焰的火焰。其特点是燃烧不完全,温度略低于化学计量焰,具有还原性,适合于易形成难解离氧化物的元素测定,但它的干扰较多,背景高。
贫燃焰──指助燃气大于化学计量焰的火焰。它的温度较低,有较强的氧化性,有利于测定易解离、易电离的元素,如碱金属。
特征浓度──在火焰原子吸收中常用特征浓度cc来表示分析灵敏度。所谓特征浓度即为能产生1%吸收(即吸光度值为0.0044)信号时所对应的待测元素的浓度(单位为μg·mL-1/1%)。
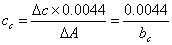
特征质量──在石墨炉中常用特征质量mc来表征分析灵敏度。所谓特征质量即产生1%净吸收(即吸光度值为0.0044)信号时所对应的待测元素的质量(单位为μg/1%)。
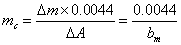
检出限──以待定的分析方法,以适当的置信水平被检出的最低浓度或最小量。常以三倍于标准偏差在灵敏度中所占的分数来表示,即
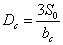 或
或 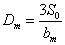
物理干扰──指样品在转移、蒸发和原子化过程中,由于溶质或溶剂的物理化学性质改变而引起的干扰。
化学干扰──指在溶液或原子化过程中待测元素与其他组分发生化学反应而使其原子化效率降低或升高引起的干扰。
电离干扰──是待测元素在形成自由原子后进一步失去电子,而使基态原子数减少,测定结果偏低的现象。
光谱干扰──是指与光谱发射和吸收有关的干扰效应。主要包括非共振线干扰和背景吸收等。
非共振线干扰──在光源发射待测元素多条谱线时,通常选用最灵敏的共振线作为分析线。若分析线附近有单色器不能分离掉的待测元素其他特征谱线,它们将会对测量产生干扰。
背景吸收──是一类特殊的光谱吸收,它包括分子吸收和光散射引起的干扰。
4.2概述
利用原子吸收分光光度计测量待测元素的基态原子对其特征谱线的吸收程度来确定物质含量的分析方法,称为原子吸收光谱法(atomic absorption spectrophotometry,AAS)或原子吸收分光光度法,简称原子吸收法。
AAS优点:
(1)检出限低 FAAS的检出限可达ng·mL-1,GFAAS可达10-14~10-13g。(FAAS如Cu为8 ng·mL-1、Cd 1ng·mL-1、Zn 1ng·mL-1、Mg 0.1ng·mL-1),(GFAAS如Cu为6×10-13 g、Cd8×10-14 g、Zn3×10-14 g、Mg4×10-14 g)。
(2)选择性好 AAS是基于待测元素对其特征光谱的吸收,因此干扰较少,易于消除。
(3)精密度和准确度高 由于原子吸收程度受外界因素的影响相对较小,因此一般具有较高的精密度和准确度。
(4)测定元素多 元素周期表中能够用AAS测定的元素多达70多种。
(5)需样量少、分析速度快 一次测定,只需10~30μL(石墨炉法)到几mL(火焰法)样品,几秒钟便可测定一个样品。
传统AAS的不足之处
1.一种元素一个灯,更换不太方便。
2.标准曲线线性范围窄,不超过两个数量级。
3.样品前处理麻烦。
4.仪器设备价格昂贵。
5.对操作人员的基础理论和技术要求较高。
新型多通道原子吸收光谱法中虽然在一定程度上解决了此问题,但价格比较昂贵。另外,对多数非金属元素还不能直接测定。
4.3基本内容
一、基本原理
基态原子吸收其共振辐射,外层电子有基态跃迁到激发态而产生原子吸收光谱。原子吸收光谱位于光谱的紫外区和可见区。
原子吸收光谱法是基于待测元素的基态原子在蒸气状态对其原子共振辐射的吸收进行元素定量分析的方法。
二、原子吸收谱线的轮廓与变宽
原子吸收光谱线有相当窄的频率或波长范围,即有一定宽度。表示原子吸收线轮廓的特征量是吸收线的特征频率v0和半宽度(Δv)。原子吸收线的宽度受多种因素的影响,其中主要有自然宽度、多普勒变宽ΔvD和压力变宽。压力变宽包括洛伦兹变宽ΔvL和赫尔兹马克变宽ΔvH两种。在通常原子吸收实验条件下,吸收线的轮廓主要受多普勒变宽和洛伦兹变宽影响。对于火焰原子吸收,ΔvL为主要变宽;而对于石墨炉原子吸收,ΔvD为主要变宽。
三、原子吸收谱线的测量
原子吸收谱线的测量包括积分吸收和极大(峰)值吸收。积分吸收采用的是连续光源加以单色器进行分光,再通过原子化器,测量吸收值,但积分吸收的测定非常困难。因为原子吸收线的半宽度很小,只有0.001~0.005nm。要分辨如此窄的谱线,其分辨率是现代仪器不可能达到的。因此尽管原子吸收线象早在18世纪就被发现,但一直未用于分析。直到1955年,沃尔什(Walsh)提出以“峰值吸收”来代替“积分吸收”。从此,积分吸收难以测量的困难得以间接的解决。峰值吸收采用发射线半宽度比吸收线半宽度小得多的锐线光源,这样就不需要用高分辨率的单色器,而只要将其与其他谱线分离,就能测出吸收值。
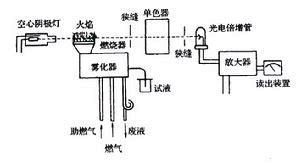
四、原子吸收光谱仪
原子吸收光谱仪又称原子吸收分光光度计,由光源、原子化器、单色器和检测器等四部分组成。
光源的作用是发射待测元素的特征共振辐射,为了测定待测元素的峰值吸收,必须采用待测元素制成的锐线光源。常用的光源有空心阴极灯、蒸汽放电灯和无极放电灯。
原子化器的作用是将样品中的待测元素转化为基态原子,以便对光源的特征谱线进行吸收。原子化器目前主要有火焰原子化器、石墨炉原子化器和低温原子化器三类。
分光系统主要有入射狭缝、反射镜、色散元件和出射狭缝等组成。在固定空心阴极灯光源原子吸收光路中,分光系统的分辨能力取决于色散元件的色散率和狭缝宽度。光谱通带(即通过单色器出射狭缝的光束的波长宽度)等于狭缝宽度与色散率倒数的乘积。W=D·S。对于具体的仪器来说,色散元件的色散率已固定,此时的分辨率仅与仪器的狭缝宽度有关。减小狭缝宽度,有利于提高分辨率、消除干扰谱线。但狭缝宽度太小,会导致透过光的强度减弱,分析灵敏度下降。因此在实际测量中要选择一适当的狭缝宽度进行测量。
检测系统有光电转换器、放大器和显示器组成,它能把单色器分出的光信号转换为电信号,经放大后以透光率或吸光度的形式显示出来。
五、测量条件的选择
分析线、灯电流、狭缝宽度、燃烧器高度、增益、样品用量。
六、原子吸收光谱法的分析方法
标准曲线法、标准加入法。
七、干扰及消除方法
(1)物理干扰 样品黏度、表面张力使样品进入火焰的速率或喷雾效率改变引起的干扰。可通过配制与样品具有相似组成的标准溶液或采用标准加入法来消除。
(2)化学干扰 指在溶液或原子化过程中待测元素与其他组分发生化学反应使得待测元素原子化效率降低或升高引起的干扰。由于产生化学干扰的原因比较复杂,消除干扰要根据具体情况采取相应的措施。消除方法是:①加入释放剂;②加入保护剂;③化学分离。
(3)电离干扰 在高温条件下,原子会电离,使基态原子数减少,吸光度下降,这种干扰称为电离干扰。消除电离干扰的方法是加入过量的消电离剂。在相同条件下消电离剂首先电离,产生大量的电子,抑制待测元素的电离。
(4)光谱干扰 非共振线干扰:在光源发射的待测元素多条谱线中,通常选用最灵敏的共振线作为分析线。若分析线附近有单色器不能分离掉的待测元素其他特征谱线,它们将会对测量产生干扰。改善和消除这种干扰的办法是减小狭缝宽度。
背景干扰:主要来自原子化器,包括蒸气中气态分子对光的吸收(无机酸、气体燃烧等)及高盐度颗粒的散射干扰。一般采用仪器校正背景的方法,有空白校正法、氘灯校正法、塞曼效应校正法等。
空白校正法──配制一个与待测样品组成、浓度相近的空白溶液,则这两样溶液的背景吸收大致相同,测得待测溶液的吸光度减去空白溶液的吸光度即为待测溶液的真实吸光度。
氘灯校正法──同时使用空心阴极灯和氘灯两个光源交替通过原子化器。当氘灯这种连续光源照射待测元素时,待测元素的共振吸收对于总入射光强度是可以忽略不计的,因此连续光源的吸光度值即为背景吸收。将锐线光源吸光度值减去连续光源吸光度值,即为校正背景后的待测元素的吸光度值。
塞曼效应校正法──塞曼效应是指在磁场作用下简并的谱线发生分裂的现象。塞曼效应校正法是磁场将吸收线分裂为具有不同偏振方向的分线,利用这些分裂的偏振分线来区别待测元素和背景的吸收,并扣除背景吸收。
4.4思考题与习题解答
1. 影响原子吸收谱线宽度的因素有哪些?其中最主要的因素是什么?
答:影响原子吸收谱线宽度的主要因素有自然宽度、多普勒变宽ΔvD和压力变宽。压力变宽包括洛仑兹变宽ΔvL和赫尔兹马克变宽ΔvH两种。对于火焰原子化吸收,ΔvL为主要变宽;而对于石墨炉原子化吸收,ΔvD为主要变宽。
2. 通常为什么不用原子吸收光谱法进行物质的定性分析?
答:由于积分吸收很难测出原子吸光度的大小,必须使用能包含待测原子所能吸收特征谱线的锐线光源,才能测准吸光度的大小。所以释放特征谱线的原子与待测物质必须是同类原子,即选用锐线光源时需知道待测原子的类型,故不能用于定性分析当中。
3. 原子吸收光谱法,采用极大吸收进行定量的条件和依据是什么?
答:为了使通过原子蒸气的发射线特征频率恰好能与吸收线的特征频率相一致,通常用待测元素的纯物质作为锐线光源的阴极,使其产生发射,这样发射物质与吸收物质为同一物质,产生的发射线与吸收线特征频率完全相同,可以实现峰值吸收。而且在特定条件下,吸光度与待测元素的浓度呈线性关系,这是采用峰值吸收进行定量的依据。
4. 原子吸收光谱仪主要由哪几部分组成?各有何作用?
答:原子吸收光谱仪主要由光源、原子化器、分光系统和检测系统这四大部分组成。原子吸收光源的作用是发射待测元素的特征谱线。为了测定待测元素的峰值吸收,必须采用待测元素制成的锐线光源;原子化器的作用是将样品中的待测元素转化为基态原子,以便对光源发射的特征光进行吸收;分光系统的作用就是将待测元素的分析线与干扰线分开,使检测系统只能接受分析线;原子吸收检测系统的作用就是把单色器分出的光信号转换为电信号,经放大器放大后以透光率或吸光度的形式显示出来。
5. 使用空心阴极灯应注意什么?如何预防光电倍增管的疲劳?
答:使用空心阴极灯时应选择合适的灯工作电流。增大灯的工作电流,可以增加发射强度,但电流过大,会使灯的阴极温度上升,多普勒效应增强,谱线变宽;严重时会使阴极溅射过强,灯内基态原子浓度过大,产生自吸,导致灵敏度下降,灯的寿命减小。空心阴极灯的电流也不能太小,否则发光不稳定,谱线强度过低。
长时间使用或辐射光强度过大,外加电压过大都会造成光电倍增管疲劳。因此应尽量避免非信号光照射和长时间无间隙使用,并尽量不用过高的电压,以确保光电倍增管的良好工作特性。
6. 与火焰原子化相比,石墨炉原子化有哪些优缺点?
答:与火焰原子化器相比,石墨炉原子化器的原子化效率高,气相中基态原子浓度比火焰原子化器高数百倍,且基态原子在光路中的停留时间更长,因而灵敏度高得多,特别适用于低含量样品分析,取样量少,能直接分析液体和固体样品。但石墨炉原子化器操作条件不易控制,背景吸收较大,重现性、准确性均不如火焰原子化器,且设备复杂,费用较高。
7. 光谱干扰有哪些,如何消除?
答:光谱干扰主要有非共振线干扰和背景吸收两类,消除非共振线干扰的办法是缩小狭缝宽度。背景吸收的消除常有空白校正、氘灯校正和塞曼效应校正等几种方法。
8. 简述原子吸收光谱法比原子发射光谱法灵敏度高、准确度高的原因。
答:激发态原子数受温度的影响大,而基态原子数受温度影响小,所以原子吸收光谱法的准确度优于原子发射光谱分析法,基态原子数远大于激发态原子数,因此原子吸收光谱法的灵敏度高于原子发射光谱分析法。
9. 背景吸收是怎样产生的?对测定有何影响?如何扣除?
答:背景吸收是一类特殊的光谱吸收,它包括分子吸收和光散射引起的干扰。分子吸收是指样品在原子化过程中,生成某些气体分子、难解离的盐类、难熔氧化物、氢氧化物等对待测元素的特征谱线产生的吸收而引起的干扰。光散射是指原子化过程中产生的固体颗粒,光路通过时对光产生散射,使被散射的光偏离光路,不为检测器所检测,测得的吸光度偏高。
扣除方法一般采用仪器校正背景的方法,有空白校正法、氘灯校正法、塞曼效应校正法等。
10. 比较标准加入法与标准曲线法的优缺点。
答:标准曲线法的优点是大批量样品测定非常方便。但不足之处是对个别样品测定仍需配制标准系列,手续比较麻烦,特别是遇到组成复杂的样品测定,标准样的组成难以与其相近,基体效应差别较大,测定的准确度欠佳。
标准加入法的最大优点是可最大限度的消除基体影响(但不能消除背景吸收)。对批量样品测定手续太繁,不宜采用,对成分复杂的少量样品测定和低含量成分分析,准确度较高。
11. 原子吸收光谱仪三档狭缝调节,以光谱通带0.19nm、0.38nm和1.9nm为标度,对应的狭缝宽度分别为0.1mm0.2mm和1.0mm,求该仪器色散元件的线色散率倒数;若单色仪焦面上的波长差为2.0nm﹒mm-1,狭缝宽度分别为0.05mm、0.1mm、0.2mm和2.0mm四档,求所对应的光谱通带各为多少?
解:由 得
得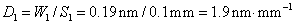
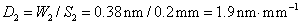
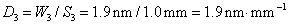
因此该仪器的色散元件的线色散率倒数为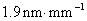
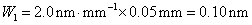
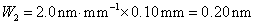


12. 测定植株中锌的含量时,将三份1.00g植株试样处理后分别加入0.00mL、1.00 mL、2.00 mL 0.0500mol﹒L-1ZnCl2标准溶液后稀释定容为25.0 mL,在原子吸收光谱仪上测定吸光度分别为0.230、0.453、0.680,求植株试样中锌的含量(3.33×10-3g﹒g-1)。
解:根据题中数据得
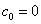
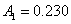
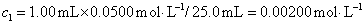
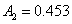
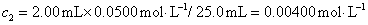
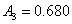
以锌标准溶液浓度对吸光度作图得图4-1,由标准曲线上查得
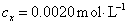
样品中锌的质量分数为w,则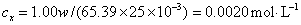
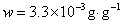
答:植株样品中锌的含量为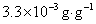 (或0.33%)
(或0.33%)
13. 用原子吸收法测定钴获得如下数据:

(1).绘制A―c标准曲线;
(2)某一试液在同样条件下测得T=20.4%,求其试液中Co的质量浓度。
解:(1)先将T%换算为吸光度A: 由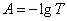 得
得

以浓度为横坐标,A为纵坐标,绘制A―c标准曲线,见图4-2。
(2)由标准曲线查得在A=0.690处对应的钴的浓度为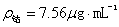 。
。
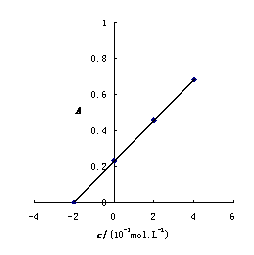
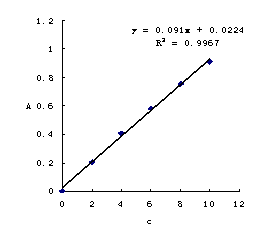
图4-1 图4-2
-
化工原理实验报告吸收实验
广西大学实验报告姓名专业月实验内容吸收实验指导教师一实验名称吸收实验二实验目的1学习填料塔的操作2测定填料塔体积吸收系数KYa三实…
- 实验报告_吸收实验
-
吸收实验实验报告
广西大学实验报告姓名专业月实验内容吸收实验指导教师一实验名称吸收实验二实验目的1学习填料塔的操作2测定填料塔体积吸收系数KYa三实…
-
填料塔吸收实验报告
实验6填料吸收塔实验报告第四组成员王锋郑义刘平吴润杰一实验名称填料吸收塔实验二实验目的1了解填料吸收塔的构造并实际操作2了解填料塔…
-
吸收实验报告
吸收实验专业环境0901学号姓名一实验目的1了解填料吸收塔德基本构造吸收过程的基本流程及操作2掌握吸收总传质系数Kya的测定方法二…
-
激光拉曼光谱实验报告---近代物理实验
激光拉曼光谱实验报告学号20xx11141054姓名牟蓉实验日期20xx328指导老师杨国建摘要本实验研究了用半导体激光器泵浦的N…
-
实验31 原子发射光谱观测分析(实验报告)
实验31A原子发射光谱观测分析实验目的1学会使用光学多通道分析器的方法2通过对钠原子光谱的研究了解碱金属原子光谱的一般规律3加深对…
-
圆二色光谱实验报告
圆二色光谱实验一实验目的1了解圆二色CD光谱的原理和使用方法2学会用圆二色光谱检测蛋白质二级构象的基本原理和方法并学会分析物质的手…
-
氢原子光谱实验报告
氢原子光谱摘要本实验用光栅光谱仪对氢原子光谱进行测量测得了氢原子光谱巴尔末线系的波长求出了里德伯常数最后对本实验进行了讨论关键词氢…
-
氢、氘光谱实验报告
实验一A氢氘光谱实验目的要求1测定氢原子与氘原子的巴耳末系发射光谱的波长和氢原子与氘原子的里德伯常数2了解WGD8A型组合式多功能…
-
氢原子光谱实验报告_-_完成版
南京理工大学大学生物理实验研究论文氢原子光谱中文摘要本实验用三棱镜对汞原子光谱进行测量得出定标曲线再对氢原子光谱进行测量测得了氢原…