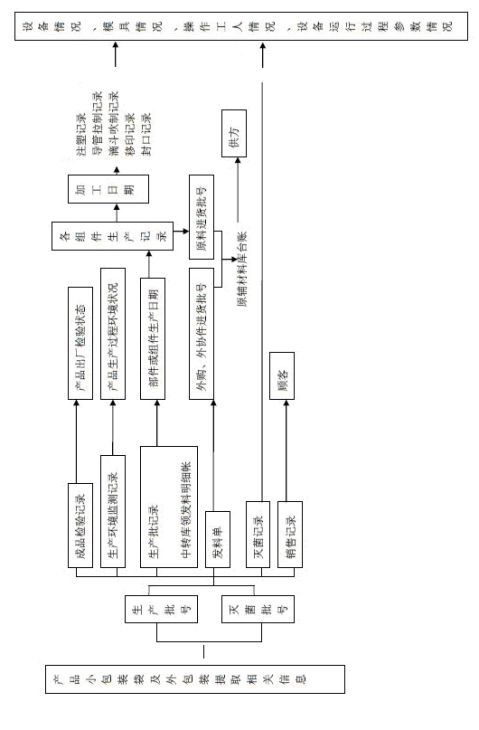
可追溯性要求及方法
可追溯性要求及方法:
①当检验过程中出现重大不合格时应进行追溯。追溯流程为:(不合格产品或部件的生产批记录)以生产日期为主线→相应生产记录或灭菌记录→生产或检验记录/环境监测记录→领发料记录→进货入库记录→进货质量检验记录→供方。 ②当顾客对产品质量投诉、退货或发生质量纠纷时应进行追溯。追溯流程为:产品小包装或外包装提取的信息(生产批号/灭菌批号/生产组号或其它可利用标记)以生产日期为主线→成品检测记录/生产记录/灭菌记录→生产检验记录/环境监测记录→领发料记录→进货检验记录→供方。
③发现产品潜在不合格时,除追溯外还应按《通告和医疗器械追回管理制度》及时追回。追溯流程为:生产批号/灭菌批号→销售记录→各产品流入单位。
产品追溯流程图
召回
1、召回分级
按照对人体可能造成伤害程度,缺陷医疗器械的召回分为三级:
一级召回:使用该医疗器械可能或者已经引起严重健康危害的;
二级召回:使用该医疗器械可能或者已经引起暂时的或者可逆的健康危害的; 三级召回:使用该医疗器械引起危害的可能性较小但仍需要召回的。
医疗器械生产企业应当根据召回分级与医疗器械销售和使用情况,科学设计召回计划并组织实施
2、召回计划
实施缺陷医疗器械召回应制订具体召回计划。召回计划应当包括以下内容:
2.1医疗器械生产销售情况及拟召回的数量;
2.2召回措施的具体内容,包括实施的组织、范围和时限等;
2.3召回信息的公布途径与范围;
2.4召回的预期效果;
2.5医疗器械召回后的处理措施。
3、召回认定
医疗器械存在以下之一情况者,为缺陷医疗器械,应当予以召回:
3.1不符合有关医疗器械国家标准、行业标准或注册产品标准的;
3.2发生医疗器械不良事件,并经过分析确认,因设计、制造上的缺陷造成或者可能造成人体伤害的;
3.3经检测、实验和论证,在特定条件下仍可能引发人体伤害的;
3.4其它不符合国家医疗器械法规、规章需要召回的;
3.5其它可能引起顾客抱怨的较轻缺陷,如规格、型号、数量或发送错误需召回的。
4、召回时限
对一、二、三、四类缺陷作出医疗器械召回决定的,管理者代表应当根据召回分级与医疗器械销售和使用情况,科学设计召回计划并组织实施。一级召回在1日内,二级召回在3日内,三级召回在7日内,应由管理者代表完成召回通知交总经理批准后由销售部采用适当的渠道和方式通知到有关医疗器械经营企业和使用单位。五类缺陷由销售部相关销售员提出申请,经质检部验证后如确系质量
问题的,由总经理批准同意召回。
5、召回通知内容
一、二、三、四类缺陷的医疗器械召回通知至少应当包括以下内容:
5.1这一产品可能有问题,需要召回;
5.2尚未销售和使用的所有产品必须立即停止销售和使用;
5.3接受医疗器械生产企业委托的负责直接销售的经营企业,应当将召回通知转发到被召回产品所在的其他单位(如果有需要)。
5.4对被召回的医疗器械如何处理的建议。
6医疗器械召回上报程序
6.1医疗器械生产企业做出医疗器械召回决定的,应当立即书面告知所在地省、自治区、直辖市药品监督管理部门,并且在5日内填写《医疗器械召回事件报告表》,将调查评估报告和召回计划同时提交给所在地省、自治区、直辖市药品监督管理部门备案。
6.2在实施召回的过程中,根据召回计划定期向药品监督管理部门提交召回计划实施
情况报告报告召回计划实施情况。
7召回产品处理
7.1管理者代表应做好对召回医疗器械的处理产生的详细的记录。
7.2必须销毁的医疗器械,应当在药品监督管理部门监督下销毁。
7.3对可以通过采取警示、检查、重新标签、修改说明书、替换、销毁等方式消除其危害的产品,可以在产品所在地完成上述行为。
7.4质量问题召回的产品,进厂后,必须隔离存放,做待检标识,经质检部核实,结果为不合格的,由销售部根据《不合格品控制程序》做出处理,必要时采取纠正和预防措施。
7.5在召回完成后,应当对召回效果进行评价,向药品监督管理部门提交医疗器械召回总结报告。
第二篇:可追溯性医疗器械不良反应管理方法的应用效果分析

-
20xx年重整变电所“党员示范岗工作总结”
党员示范岗工作总结为贯彻落实科学发展观,加强党员队伍建设,我重整变电所在上级党支部的指导下,精心开展了“党员示范岗”创建活动,在全…
-
在外企工作的人的总结
1、任何的工作一定要从前到后全部做完,如果我只让你做1,你要自己想想这个事儿有没有2和3,如果我没有时间教你全部的过程,请你自己多…
-
班委工作总结
软件1105班委工作总结光阴似箭,岁月如梭,转眼间,大二下学期已经告一段落,作为班级的领头者与服务者,在此对于这学期的工作做如下总…
-
个人总结(党员)
20xx年个人总结(党员)本人xxx,参加了20xx年x月xxx卫生局公招考试,荣幸又成为了一名基层医务工作者。虽然我刚到一个新的…
-
一届二次教代会提案工作总结
一、基本情况一届二次教代会共征集代表提案176件,提案人和附议人687人次,提出提案单位38个。内容分为发展规划、学科建设、学校管…