医疗器械工艺用气验证报告
工艺用压缩空气验证报告
一、基础
在一次性使用医疗器械生产中,工艺用压缩空气主要用于软管挤塑型、滴斗吹塑成型以及检测一次性使用输液(血)器、一次性使用静脉输液针、药液过滤器的气密性和畅通性,其直接接触产品的内表面,为了严格控制工艺用压缩空气中微粒和微生物对产品造成的污染,保证产品使用医疗器械生产的要求。
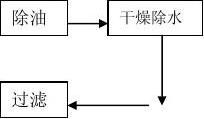
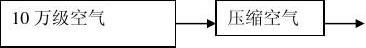
二、工艺用压缩空气的供气工艺流程及设备
1、设备(主要技术参数见附件1)
1.1 W-0.9/8型空气压缩机
1.2 JH-1/1型压缩空气除油器
1.3 压缩空气冷冻式干燥机
1.4 OYJ-A系列压缩空气组合净化器
2、工艺流程
三、工艺用压缩空气洁净度的验证
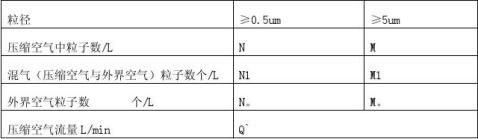
1、工艺用压缩空气洁净度要求

使用的仪器、设备、材料
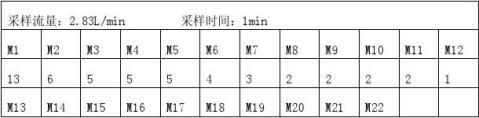
09-9型激光粒子计数器(采样流量2.83L/min)
JYQ浮游菌采取器(采样流量50 L/min)
热球式电风速计
PG/2007恒温/干燥箱


电子数显卡尺
Φ150mm真培养皿
无菌普通肉汤固体培养基
3%双氧水、75%酒精
洁净、无菌的SS-50外套、二通、Φ3.0PVC导管,线型药液注射伯
3方法
3.1取样点的设置
将线型药液注射伯与压缩空气气咀及二通连接,再用Φ3.0PVC导管连接二通及SS-50外套的6:100外圆锥接头。取样点在SS-50外套内。
3.2测压缩空气流量
3.2.1测SS-50外套内径
用电子数显卡尺取5个不同方位测内径。
3.2.2测SS-50外套出口处压缩空气流速
接通压缩空气电磁阀,使压缩空气完全开放,用热球式电风速计测SS-50外套出口处压缩空气流速。
3.2.3计算压缩空气流量
Q`= ∏ ·(D/2)2·V X 60 X 10-3
式中: Q`---压缩空气流量 L/min
D---SS-50外套内径mm
V-- SS-50外套出口处风速m/s
3.3测尘埃粒子数(依据GB/T 16292-1996)
3.3.1接通激光粒子计数器,使其预热泪盈眶15min,调粒径为0.3um,待自净完毕,将取样连接管与粒子计数器进气口连接,再将采样口套于SS-50外套内,接通压缩空气电磁阀,使SS-50外套内完全充满空气调粒径,开启粒子计数器流量阀,分别测0.5um和5um粒子数。
3.3.2计算单位体积粒子数
当Q﹥2.83L/min时,N= (1/nΣNi)÷2.83(i =1……n)
M= (1/nΣMi)÷2.83(i =1……n)
式中:N:0.5um单位体积粒子数 个/L
M:5um单位体积粒子数 个/L
Ni:2.83压缩空气中测得的0.5um粒子数 个
Mi:2.83压缩空气中测得的5um粒子数 个
n:测量次数
当Q`﹤2.83L/min时, N= N1-N。/2.83(2.83- Q`)
M= M1-M。/2.83(2.83- Q`)
3.4测工艺用压缩空气温、湿度
3.5测浮游菌(依据GB/T16293-1996)
3.5.1培养基的准备及灭菌
3.5.2仪器的消毒处理
用3%双氧水消毒浮游菌细菌采样器外表面、采样器顶盖、转盘罩子内外表面、采样口以及采样管。
3.5.3采样程序
将取样口套于SS-50外套内,接通压缩空气电磁阀,使SS-50外套内充满压缩空气。 开真空泵抽气10min,使残留在仪器内消毒剂蒸发并调整流量与转盘速度。
关真空泵,放入培养皿,盖上盖子后调节采样器逢隙高度 。
选定采样时间测试。
3.5.4培养
30℃~35℃培养48h,并选3只空白平板对照培养。
3..5.5计数
用透射光于培养皿背面或正面照射,肉眼观察、标记,然后用5~10倍放大镜查是否有遗漏。若有2个或2个以上菌落重叠,可分辨时,仍以2个或2个以上菌落计数。
3.5.6计算单位体积浮游菌浓度
当Q`﹥50L/min 时,C=(1/nΣCi)10?÷50(i =1……n) 个/M3
式中:C—单位体积压缩空气中浮游菌浓度 个/M3 Ci—以50L/min流量采样5min浮游菌 个 当Q`﹤50L/min 时,C=C1-C0/50(50- Q`) 个/M3
式中 C 、C1 、C0分别表示压缩空气、混气(压缩空气与外界空气)、外界空气中单位体积浮游菌数 个/M3。 3. 6结果


3.6.3计算压缩空气流量 Q`=∏ ·(D/2)2·V X 60 X 10-3
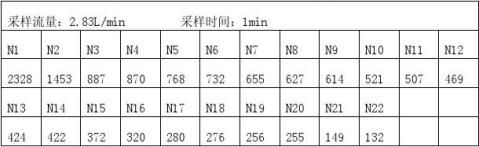
利用Grubbs检验法对以上数据极端值进行取舍 a.n=22 N(平均)=604.5 S=486.97
T=
查Grubbs检验临界值表 T(22,5.0%)=2.60 因T﹥T(22,5.0%)所以N1=2328为异常,弃去. b.n=21 N=522.43 S=305.6 T=
查Grubbs检验临界值表 T(21,5.0%)=2.58 因 T﹥T(21,5.0%)所以N2=1435为异常值,弃去。 c.n=20 N=476.8 S=228.68
T= 或T=
查Grubbs检验临界值表 T(20,5.0%)=2.56 因 T﹤T(20,5.0%)所以无异常值。
综合分析N1=2328 N2=1435为异常值,弃去。
3.6.5大于0.5um粒径粒子数
因为Q`=159.4L/min,大于采样流量2.83L/min 所以N=

、利用Grubbs检验法对上组数据极端值处理 a.n=22 M=2.55 S=2.97 T=
查Grubbs检验临界值表 T(22,5.0%)=2.60 因T﹥T(22,5.0%)所以M1=13 为异常值,弃去。 b.n=21 M=2.05 S=1.88
T= 或T=
查Grubbs检验临界值表 T(21,5.0%)=2.58 因 T﹤T(20,5.0%),故无异常值。 综合分析M1=13为异常值。 3.6.7计算大于5um粒径粒子数
因Q`=159.4L/min,大于采样流量2.83L/min 所以M=
3.6.8压缩空气温度T=20℃,湿度49RH%.
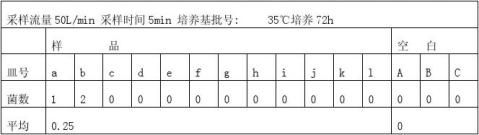
3.6.10计算浮游菌浓度
因Q`=159.4L/min,大于采样流量50L/min
所以C=


表6显示工艺有压缩空气超过一万级洁净空气要求,符合工艺要求。
产品验证
4.2使用仪器、设备、材料
PJ-Ib型微粒检测仪
水平式无菌超净台
细菌培养箱
霉菌培养箱
高压灭菌锅
无菌培养基、胰酪胨大豆、固体培养基、蛋白胨水、霉菌培养基
氯化钠注射液]
无菌平皿、吸管、试管、PH试纸、放大镜、洒精灯、量筒、过滤装置
一次性使用输液器IS-V4
4.3方法(依据BS EN 1174-1: 1996、GB15980-1995、GB7918.2-1995
4.3.1供试液制备
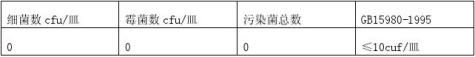
随机抽取10件样品,每件样品以无菌操作注入10ml蛋白胨水,以重力反复冲洗5次,分别分装于无菌试管中备用。
4.3.2倒平板
每样取5份平行样,用无菌吸管吸取8ml供试液分别注入8只平皿内,每皿1ml,将熔化并冷至45-50℃的胰酪胨大豆固体培养基和霉菌培养基分别倾注5只和3只平皿内,转动平皿使

样品与培养基混合均匀,等琼脂凝固后,倒置培养。 4.3.3培养
细菌于恒温培养箱中35-37℃培养3天,霉菌于霉菌/湿热培养箱中20-25℃培养5天。 4.3.4菌落计数方法
a.先用肉眼观察、标记,再用5-10倍放大镜检查,以防遗漏; b.若所有的平皿均无菌生长,报告数为每毫升小于10个。
表7显示,初始污染菌为0,据GB15980-1995,判初始污染菌≤10cuf/皿,符合GB15980-1995要求。 4.4微粒污染 4.4.1抽样
随机抽取8套一次性使用输液器IS-V4,除去药液过滤器。 4.4.2操作步骤(GB8368-1998) 6.1附录) 4.4.3测试结果
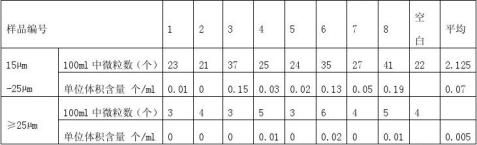

4.4.4结论
表8显示,粒径15?m-25?m微粒污染数0.07个/ ml,符合≤1个/ ml的要求,粒径≥25?m微粒污染数0.005个/ ml,符合≤0.5个/ ml的要求,因此,产品微粒污染符合GB8368-1998 6.1要求。 5.结论
表9显示,使用该工艺用压缩气体生产的产品符合GB15980-1995和GB8368-1998 6.1要求。 工艺用压缩空气的监控与检查
PD/BF/115《工艺用压缩空气管理制度》规定了工艺用压缩空气技术要求及监测频次,以确保供所质量符合一次性使用医疗器械产品质量的要求。 制气设备的维护保养
6.1 ED/BF/014《设备管理制度》对设备的选型、购置、安装、维修、保养和操作及操作人员进行了规定,确保其满足生产的需要并使设备处于正常运行状态。
6.2 ED/BF/009《设备检查制度》按照设备规定的性能,对设备进行定期检查、测定,消除隐患,为维修提供必要的信息。
6.3 ED/BF/008《设备大中小修制度》对生产过程中通过状况监测出设备的异常情况,确定维修项目进行小修,据小修和设备定期检查的记录,每年一次中修,据定期检查、日常小修、、定期的中修信息和设备技术状况,计划并实施设备的大修工作,以保持设备良好的技术状态,并按规定的效率连续不断地进行正常供气。
6.4 ED/BF/013《设备三级保养检查制度》对设备实行定人定机考核上岗和三级保养。 综合分析与验证结论
7.1该工艺生产的工艺用压缩空气微粒含量,粒径大于0.5um的粒子浓度为168.5个/L,符合≤350个/L要求,粒径大于5um的粒子浓度为0.72个/L,符合≤2个/L,浮游菌1个/L,浮游菌1个/m3,符合≤100个/m3要求,相对湿度49%,符合≤65%要求,因此,工艺用压缩空气符合用气工艺技术要求。
7.2使用该工艺用压缩空气的产品,其初始污染菌小于10个cfu/件,微粒污染:粒径在15um~25um含量为0.07个/ml,符合≤1个/ml要求,因此,使用该工艺用压缩空气的产品符合GB8368-1998和GB8369-1998中一次性使用输液(血)器对产品的技术要求。
7.3 对工艺用压缩空气进行日常的监测和管理,并对产气设备时行定期检修、维护和保养,保证了工艺用压缩空气的供气质量及持续有效地正常运行。
综合上述,工艺用压缩空气既符合产气工艺技术要求,也符合产品使用要求,并通过对工艺用压缩空气的严格控制和管理,能够保证一次性使用医疗器械产品的安全、有效性。
工艺用压缩空气验证小组
第二篇:工艺审查验证文件及验证报告
《※※※※※※※》工艺审查验证文件
一、小组的组成
验证项目名称:《※※※※※※※》的工艺审查验证
验证小组组长:※※※(技术总监)
验证小组成员:※※※(生产部经理)
※※※(质量部经理)
※※※(技术部经理)
二、验证方案
(一)验证目的:确认《※※※※※※※》的生产工艺能高度可靠并始终如一地生产出符合质量标准要求的产品。
(二)质量标准:《※※※※※※※》的各项性能指标应达到《※※※※※※※》标准的要求。
(三)验证内容
1.资料档案审核
(1)产品标准文件(包括制造和检定规程)
(2)产品使用说明书
(3)20100101、20100201、20100301批生产记录
2.检测和检测结果记录
(1) 20100101、20100201、20100301自测报告
(2)留样观察及稳定性结果分析报告
(四)审查方案的起草和审批
- 1 -
《※※※※※※※》工艺审查验证报告
20xx年10月20日至20xx年10月23日,《※※※※※※※》工艺审查小组根据批准的工艺审查方法,对该产品的工艺进行了审查,审查结果如下:
(一)资料档案的审核和归档
(二)检测结果
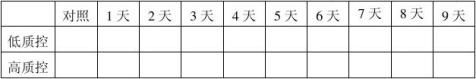
1.批号20100101、20100201、20100301自测报告(1份)。 2.留样观察及稳定性结果分析报告。(见下列表格) 20100101批成品37℃热稳定结果表:(单位:mg/ml)
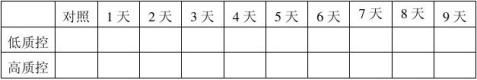
20100201批成品37℃热稳定结果表:(单位:mg/ml)
20100301批成品37℃热稳定结果表:(单位:mg/ml)
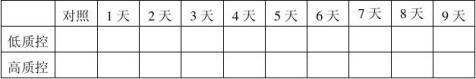
- 2 -
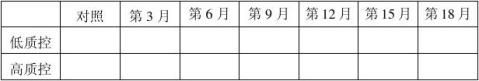
20100101批成品4℃稳定性结果表:(单位:mg/ml)
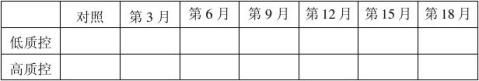
20100201批成品4℃稳定性结果表:(单位:mg/ml)
20100301批成品4℃稳定性结果表:(单位:mg/ml)
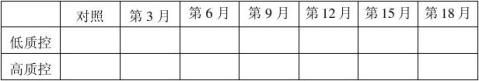
结果:经稳定性考核,产品37℃考核9天及4℃保存18个月均能达到检定要求。
(三)结论和说明
1、审查文件资料完整属实,批制检记录为生产工艺提供历史数据审查。 2、批记录比较完整,符合要求。
3、产品质量的留样观察及稳定性结果考核指标达到预期要求。 4、批批检合格率达100%,产品的生产工艺能保证生产符合国家质量标准要求的产品。
5、审查周期为一年,再审查时间为20xx年10月20日。

- 3 -
-
医疗器械行业分析报告
医疗器械行业分析报告目录一国医疗行业现状1产业结构2行业规模3产销情况4盈利情况5出口情况二中国器械产业面临的问题三医疗器械产业发…
-
医疗器械产品技术报告
医疗产品技术报告医疗器械产品技术报告审批流程如图所示医疗器械产品技术报告的要求产品技术报告应能支持产品标准安全风险分析报告临床试验…
-
医疗器械行业研究报告
医疗器械行业研究报告1目录一医疗器械行业概述311医疗器械七大类别312市场空间巨大4二医疗器械市场情况错误未定义书签21全球医疗…
- 医疗器械自查报告(完整版)
-
医疗器械调研报告
关于20xx年医疗器械行业发展调研医疗器械行业是一个快速扩容的新兴市场据国家统计局的统计20xx年我国医疗器械行业规模以上企业收入…
-
医药医疗器械质量安全自查报告
医疗器械质量安全自查报告在食品药品监督管理部门的监督指导下,我公司认真履行《质量承诺责任书》中所作的承诺,促使质量管理体系各个环节…
-
20xx年医疗器械抽样工作情况的总结报告
20xx年医疗器械抽样工作情况总结报告XX市食品药品监督管理局:为了加强医疗器械质量监管,进一步规范药械市场秩序,保证产品使用安全…
-
医疗器械临床使用安全事件监测管理制度、报告制度及报告表
医疗器械临床使用安全事件监测管理制度为加强医院医疗器械不良反应监测管理工作依据国家医疗器械监督管理条例医疗器械不良反应事件监测和再…
-
6.9.4.1医疗器械临床使用安全事件监测管理制度、报告制度及报告表
医疗器械临床使用安全事件监测管理制度为加强医院医疗器械不良反应监测管理工作依据国家医疗器械监督管理条例医疗器械不良反应事件监测和再…
-
申请医疗器械经营许可证自查报告(上报版)
有限公司有限公司申办医疗器械经营企业许可证自查报告有限公司成立于20xx年9月20xx年9月取得药品经营许可证20xx年2月取得药…
-
医疗器械注册产品分析报告
XXXXXX产品质量跟踪报告一试产期间产品的生产销售情况1试产期间XXXXX产品生产的批次为X数量为XXX2试产期间XXXXX产品…