ϵͳ����ʵ�鱨���123
��ϵ ͳ �� �̡�
ʵ�鱨���
20 - 20 ѧ�� �� ѧ��
�� ����
ѧ �ţ�
�� ����
�ڿν�ʦ��
ʵ��ѧʱ��
��Ϣ����ϵ
20##��9��
ʵ��һ ���ӷ���
һ��ʵ��Ŀ�ģ��������ӷ���������������
�������ݣ�
1.SPSS����
2.���ӷ���
3.�������������ݼ���Դ
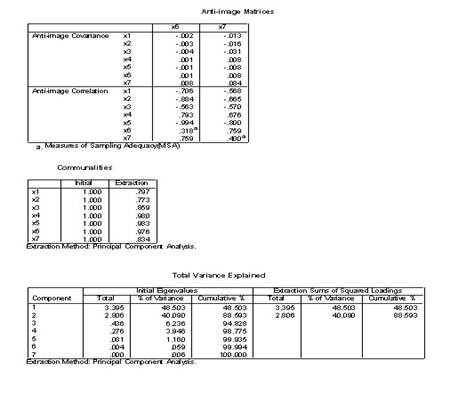
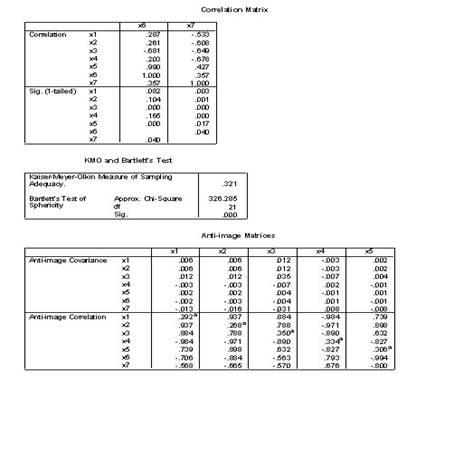
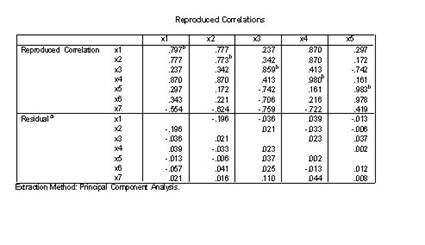
�±�����Ϊ25�������˵�7��������������7����������ָ����������ΪX1��X7����Ը����Ͻ������ӷ���
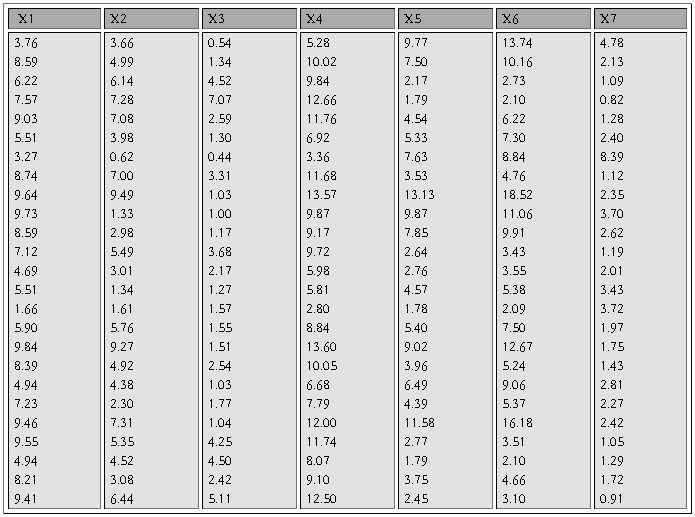
����ʵ����̼���������
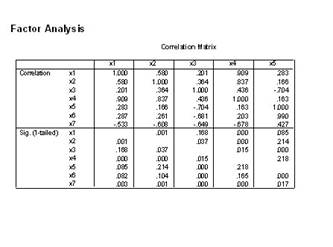
1.��Analyze��Data Reduction��Factor˳���˵�������ӷ������Ի���
2.ѡPoliticalEconomy��Calculus1 ��ComputerCulture��Microeconomics ��Algebra ��Calculus2��VBΪ���������͵��ұߵ�Variables���С�
3.�����Ի����е���Extraction��ť����Ӧ�ĶԻ����У�
1)Method�˵���ѡPrinciple components�ʹ�����ɷַ���������
2)Analyz����ѡCorrelation matrix�������ؾ���
3)Extract����ѡ��Number of factors 2
4)Display����ѡ��Unrotated factor solution����ʾδ��ת�����ӽ����ͬ��ѡ��Scree plot��Ҫ����������ֵ��ɢ��ͼ��
5)Maximum iteration convagence 25�������������о�Ϊ��������������25.
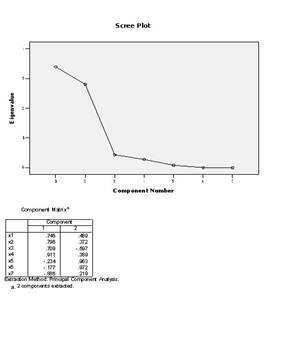
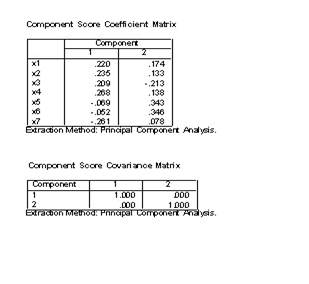
4.���Ի����е���Score��ť������Ӧ�ĶԻ�����ѡ��Save as variables
������Method����ѡ��Rregression��Ҫ��ͨ���ع鷽���������ӵ÷ֲ������ӵ÷���Ϊ�������浽�����ļ��С�
5.����Descriptives��ť���ڶԻ���Statistics����ѡ��Initial solutionѡ�
6.�����Ի����е���OK��ťִ�����㡣
�ġ��������������������
�塢ʵ���ĵ����
ʵ��� �������
һ��ʵ��Ŀ�ģ�����ģ���������������������
�������ݣ�
1��SPSS����
2���������
3���������������ݼ���Դ
29����ͯ��Ѫ�쵰�ף�g/100ml������Ԫ�أ���g/100ml���ⶨ������±���������Ԫ�صIJⶨ�ɱ��ߡ���ʱ������ϣ��ͨ�������������R��ָ����ࣩɸѡ������ָ�꣬�Ա�����ÿ�ݵ����۶�ͯ��Ӫ��״̬��
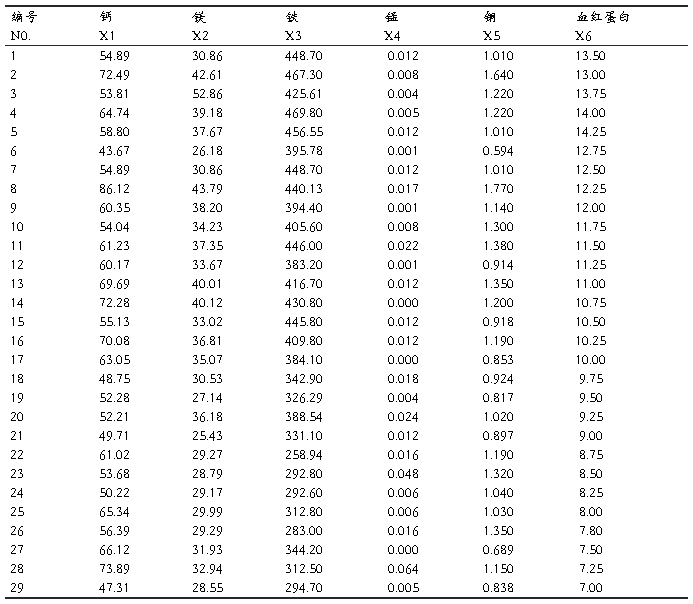
����ʵ����̼���������
1.�ȴ����������SPSS 11.5 for Windows.exe,��Variable View�и�����Ŀ�����������
2.Data View������������
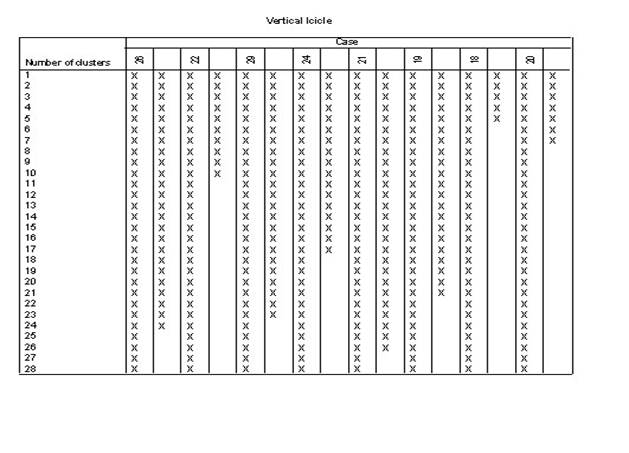
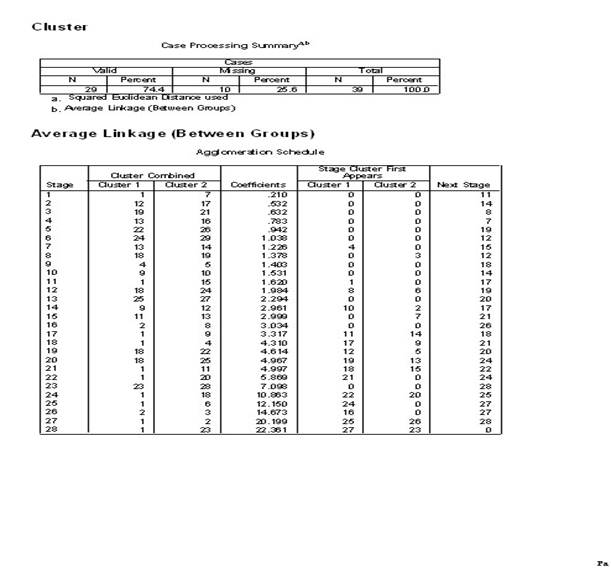
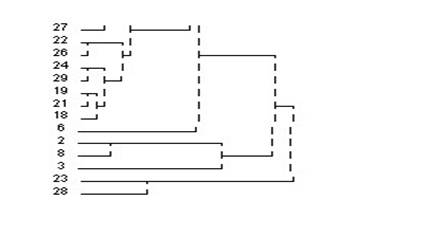
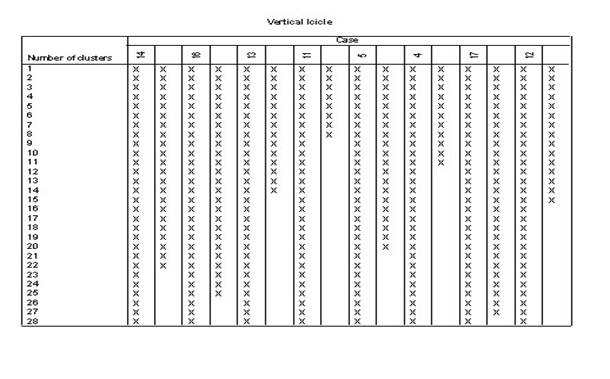
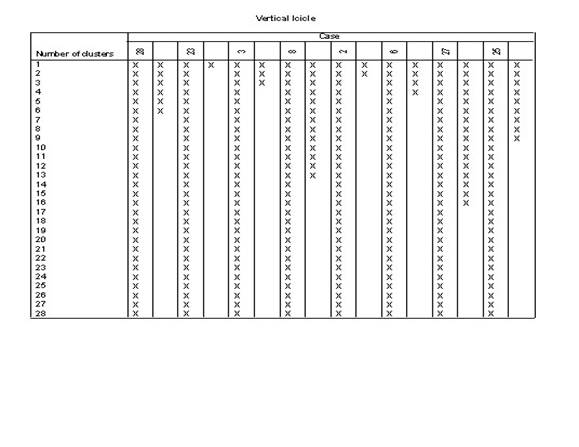
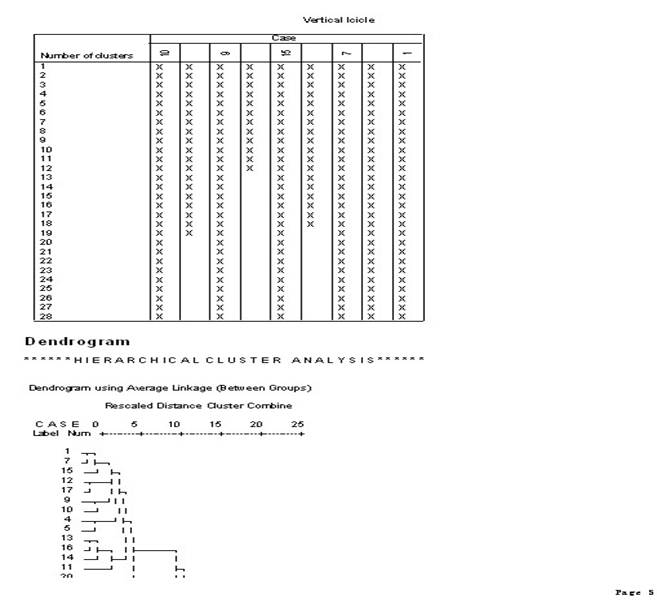
3.��������ϵͳ���෨��������ݽ��о���
4.ѡ��˵�:��Analyze������lCassify������iHerarchical Cluster����Ȼ��ѡ������ξ�������ı����������Ŀ��Եijɼ�����Variable��s�������У���ѡ��һ���ַ��ͱ�������������Ϊ��DZ�������Label Cases by�����С�
5.����Plots�������ѡ��
6.����Statistics�������ѡ��
7.����Method�������ѡ��
8.�Ե�һ��������б��棬��������Ϊ��xx.sav����ͬʱ����������
�ġ��������������������
�塢ʵ���ĵ����
ʵ���� ϵͳԤ�ⷽ���Ƚ�
һ��ʵ��Ŀ�ģ�����ϵͳԤ�ⷽ����������
�������ݣ�
1��SPSS������DPS��������
2����Ԫ���Իع���������ع顢��ɫģ��Ԥ��
������Ԫ���Իع����
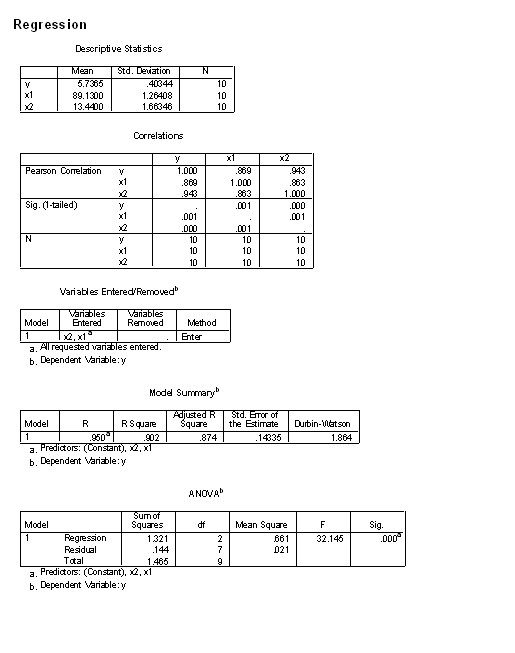
��1����Ԫ���Իع�����������������ݼ���Դ
ijҽʦ���10��3���ͯ�����ߣ�cm�������أ�kg������������cm2���������¡����ö�Ԫ�ع鷽��ȷ�������ߡ�����Ϊ�Ա�����������ΪӦ�����Ļع鷽�̡�
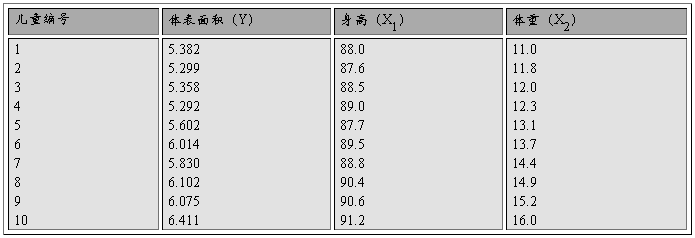
(2) ��Ԫ���Իع����ʵ����̼���������
1��Ӧ��ͳ��ѧʵ��ָ���飬�½�excel��
2����SPSS�������ݵ���
3��������ѡ��ع������ѡ������
(3)��Ԫ���Իع�����������������������
����ʵ���ĵ����
ʵ���� ���Թ滮
һ��ʵ��Ŀ�ģ��������Թ滮������������
�������ݣ�
1��WinQSB����
2�������Թ滮��������
3���������������ݼ���Դ
Ϊ�о�������Ѫѹ��Ѫ�����̴��Թ��IJ������ã�ijҽʦ�ⶨ��50-59����IJ���15����������16��������ѹ�͵��̴�ָ�꣬������£������б�����������б����Ա����ٴ�������ɸѡ���IJ��ˡ�
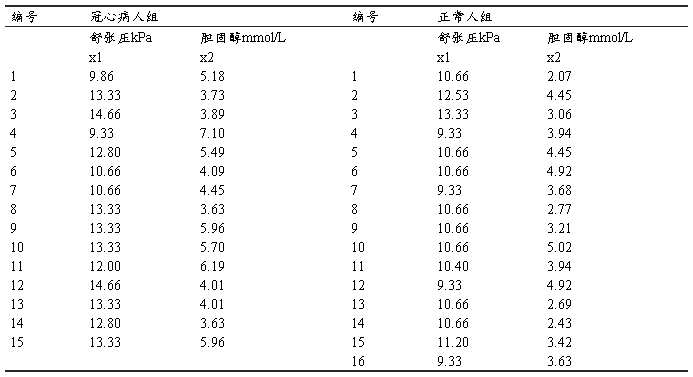
����ʵ����̼���������
1�����������ļ���
����ѹ�����̴��ı������ֱ���x1��x2��ʾ�������IJ������Ϻ����������Ϻϲ���һͬ���롣�ٶ���һ������Ϊresult���������ֹ��IJ������Ϻ����������ϣ������IJ������ϵ�resultֵ��Ϊ1�����������ϵ�resultֵ��Ϊ2��
2��ѡ��˵���Analyze��Classify��Discriminant���������Discriminant Analysis���Ի��ӶԻ������ı����б���ѡ�����result�����롰Grouping Variable����������Define Range����ť���ڵ����ġ�Discriminant Analysis: Define Range���Ի����У������б�ԭʼ���ݵ�������䣬��Minimum������1����Maximum������2��
3���ӶԻ������ı����б���ѡx1��x2��ʹ֮���롰Independents������Ϊ�б�����Ļ������ݱ���
4��������Statistics����ť��������Discriminant Analysis: Statistics���Ի����ڡ�Descriptive������ѡ��Means����Ը���ĸ������������������������ڡ�Function Coefficients������ѡ��Unstandardized�����ʾ�б̵ķDZ���ϵ����
5��������Classify����ť��������Discriminant Analysis: Classification���Ի����ڡ�Plot�� ��ѡ��Combined groups������ϲ����б����ֲ�ͼ���ڡ�Display����ѡ��Results for each case�����ԭʼ���ϸ��ݽ������б�����һ�ش����б�ͬʱѡ��Summary table��������ֻش��б��������ܽ����ۡ�
6��������Save����ť��������Discriminant Analysis: Save New Variables���Ի���ѡ��Predicted group membership������ش��б�Ľ������ԭʼ���ݿ��С�
7��������OK����ť���õ���������
�ġ��������������������
�塢ʵ���ĵ����
�ڶ�ƪ��ʵ�鱨��ϸ��123
ʵ�鱨��ϸ��123.txt�Ҷ�����۸����ˣ������ñ����۸��� һ������ô�������㼸����ʲô�Ұ�������Ҫ���ָ����Ҹ� �����Ҳ����� �������ʱ����һ��Ҫ�ҵõ��㲻�������۸�����ȫ����ֻ���Ҳſ��ԣ������㣬�±��Ӱɣ���
�л���ѧʵ��ָ����
The Guides to Experiments in Organic Chemistry
�����ã�
��ѧ�����ѧԺ ��ѧ�������
20xx��10��
1.1 �л���ѧʵ���ҹ���
��ʦ��������ȫ���豸��ȫ�Լ�ʵ��˳�����еĽǶȽ����л���ѧʵ���ҹ���Ŀ���DZ�֤�л���ѧʵ���������У�����ѧ���������õ�������⡢���������������ʵ���������ѧϰϰ�ߣ�����ѧ����ѧ��ʵ�鷽��������֤ʵ�鰲ȫ��
A�� ����ʵ���ѧ��ٺ�ʵ��������������Ԥϰ��������Ԥϰ��Ҫ��ȷʵ��Ŀ�ģ���Ϥʵ�����ݣ�����ʵ������ݲ�����ص��������ϣ���ʵ����ʵ��ǰ��һ����������������ʵ����֮ǰ������ʵ������������
B�� ������Ϥʵ���ҵ�ˮ��ú�����豸����ʵ����������ġ�����ҩ��ķ��õص��ʹ�÷�������ģ��ѵ���Ȼ�ķ�����
C�� ���ָ����ʦ��Ҫ����ʵ���������涨�IJ��衢�������Լ��Ĺ�����������ʵ�飬�����˷��Լ���ԭ���ϣ���Լˮ��ú���ȡ�
D�� ʵ�������Ҫ����۲�ʵ������ʱ��¼��ʵ����̡�ʵ�������ʵ�����ݡ�
E�� ʵ��ʱӦ���ؼ��ɣ����ְ�����ʱ������ʵ��̨��ʵ���ҵ����ࡣ
F�� ���������������豸�����豸���ߵȣ���ָ���ص�ʹ�ù����豸�ȣ���������á����࣬����������⼰ʱ��ָ����ʦ�㱨����ʱ��ȡ��ʩ��ʹ�����豸�����ܹ�������ʹ�á�
G�� ������ʵ���ҳԶ��������̡�
H�� ʵ������뿪ʵ����ʱ��Ӧ��ˮ���硢ú�����Ŵ��ȹرպã�ֵ������ɨʵ������������ָ����ʦͬ����뿪ʵ���ҡ�
I�� ��Һ��������������Ҷ����ҷţ�Ҫ�����ռ�����ͳһ������ſ����ŷźʹ��á�
1.2�л���ѧʵ���Ұ�ȫ֪ʶ
�л���ѧʵ�����õ�ҩƷ��������ȼ���ױ����ж��ģ����õ������豸�����Dz�����Ʒ��һЩ���;��ܲ���������Ϊ�˱�������Ĵ�����ɲ�Ӧ�õ��¹ʣ�����������ʵ���ҹ���Ļ����ϣ�����ѧϰʵ���Ұ�ȫ֪ʶ��ʱ�����Ӱ�ȫ���⣬��߾��裬�ϸ����ز�����̣���ǿ��ȫ��ʩ��
A������ѧϰ������ʵ���Ұ�ȫ�����мǰ�ȫʵ����ʵ��˳�����еĸ������ϣ���ʵ�������ʼ���μǰ�ȫ��һ������������ȫ������֮�ء�
B����������ʵ�����¹ʵ�Ԥ����ʩ������Ԥ�����֣�Ԥ����ը��Ԥ���ж���Ԥ������ȡ�
C�����������¹ʵĴ����ͼ��ȴ�ʩ���������ֵĴ������������ˣ����ˣ�ҩƷ���ˣ���ը���ж��ȡ�
D����Ϥ�����þߵķ��ú�ʹ�÷�����
E����Ϥ���õĻ����ȵ绰��
F���мǷ�������һ��Ҫ�侲�����Ʋ�ȡ��ʩ������������ȫ���Ʋ���ȫ���������������Ա��ͬ�����¹ʡ�
1.3ʵ��Ԥϰ��ʵ���¼��ʵ�鱨��Ļ���Ҫ��
A��ѧ��������������Ԥϰ������������ϣ�����д��Ԥϰ���棬��Ԥϰ��������Ԥϰ����д�ò�����Ҫ��IJ��ý���ʵ����ʵ�飬���Ҹ��辯��ͽ�����
B��ʵ�����һ��Ҫʵʱ����ʵ���¼����ʵ������й۲쵽��ʵ��������ʵ��ѧ�ؼ�¼���������Ҿ������˶���˼�������ۡ��������ϻ�������ʦ����Խ������۲������
C��ʵ�����������ʵ�����ݣ�����дʵ�鱨�棬ע��ʵ�鱨��Ĺ淶�ԺͿ�ѧ�ԡ����ҽ�����ۿ������������ܽ�֪ʶ���ʵ�鷽����
1.4�л���ѧʵ������
�����л���ѧʵ����صĹ����顢�ο��顢�ڿ���־����ѧ���ȣ�����ѧ���������ͼ��ݺ�������Դ���Ծ��������ף��ռ����ϣ��ܽ����ϵȡ�
�л��������������������Ʊ�ʵ�鷽����ȷ�������ᴿ�����﷽������Ҫ���ݣ�Ҳ���������л���������������ʼ����л��������ˣ�����ʹ�û�ѧ�ֲ���йزο����ѧ���л���ѧʵ������Ҫ�ġ������ڿ���ʵ���ѧ�о��Եø�����Ҫ�������ʵ���÷�Ӧ�Լ��İ�ȫ���ݻ��ò�����ۡ��е�ȶ�����ʹ�û�ѧ�ֲᣬ�������ʵ����о���ʵ����벻����ѧ�ֲᡣ�ܹ�������ʹ���ֲ�Ͳο��齫�����ȼ�����ʵ�������ѵ�ʱ�䡣����Ƽ�ʹ���л���ѧʵ����õ��ֲᡢ�ο��������¼�����
���л���ѧʵ�鳣�������ֲᡷ�������棩����������߰�������������ѧ�����磬1997��������л���ѧʵ�����ģ���������������ѧ�����л���ѧʵ�����̲ģ�ѧ��������������һ�ᡣ�л���ѧʵ�鳣�����ġ���������������������͡��л�������������������Ǹ��ֲ����Ҫ���֣��������������ƣ�Ӣ�ģ�����ѧʽ����Է�����������ɫ�;��͡�����ܶȡ��۵㡢�е㡢�۹��ʺ���ˮ���������е��ܽ���Լ���Ŀ��Դ���ڳ�������ǰ����ʹ��˵���������л�ѧʽ���������ֲ�ĵڶ��������������л���ѧʵ���йص�����ѧ��������ݣ�����ˮ�ı�������ѹ�����л������ڲ�ͬ�¶����ܽ�ȣ������������л��ܼ�
�е��ܽ�ȣ��л���������ܽ�ȣ���ҺŨ��������ܶȣ�ˮ��Һ������ѹ��ˮ��Һ�����¶ȣ�ˮ��Һ�����¶ȣ��л����Ľ��볣���ȡ��ֲ�ĵ������֣��������ʵİ�ȫ���ݣ����磬��ȼ���ʵ�����ͱ�ը���ޣ�������ijЩ��ѧ���ʵ�����Ũ�ȵȡ�
��Handbook of Chemistry and physics����������ѧ��˾����ģ��ּ��CRC�ֲᡣ
��Merck Index������һ���л���ѧʵ���ҳ��õIJο��飬��Ҫ����ҩ���Ȼ�������һЩ�������л������
�л���������Ϣ�ĵ�������Դ�ǹ�Ӧ�̵Ļ�ѧ�Լ�Ŀ¼������ְ汾�ġ���ѧ�Լ�Ŀ¼������������ƷĿ¼������������Ӧ���Լ�����Ʒ��������ѧ���ʼ�ʹ�ð�ȫ�Եȡ�
�л���������Ϣ�ĵ��ĸ���Դ�����������ɸ��ݸ�ʵ���ҵ���������ѧ��վȥ���һ�ѧ���ϡ�
2.1���ò���������ʵ��װ��
���л�ʵ���о������õ�һЩ����������ʵ��װ�ã���Ϥ��Щ������װ�ü���ά��������ʮ�ֱ�Ҫ�ġ�
2.1.1��������
���õIJ���������Ϊ���࣬��ͨ���������ͱ�ĥ��������
��ĥ�ڣ�����˼�壬�ӿڲ�λ�ijߴ��С����ͳһ�ģ��������ġ����磬14�ڡ�19�ڡ�24��ָ�ľ���ĥ�ڵ�����ֱ���ֱ�Ϊ 14 mm��19 mm�� 24 mm��ֻҪ����ͬ�ߴ�ı�ĥ�ڣ��֮������װ���Ǻϡ��Բ�ͬ�ߴ��ĥ��������������ͨ����Ӧ�ߴ�Ĵ�Сĥ�ڽ�ͷʹ֮����ӡ�
��ʹ�ñ�ĥ�������Ĺ�����Ӧ��ע�⣬װ��ʱҪ���룬�����������ͣ��������ѡ�һ������£�ĥ�ڴ�����Ϳ����������ѹ����ʱ��Ӧ�ʵ���ͿĨ���֬��ʵ�������Ӧ��ʱ��ж����������ճ����ж�����ñ�ĥ����������������������ͼ2-1��ͼ2-3��
����ͨ����������ȣ���ĥ������Ҫ��ö࣬���������ڱ�ĥ��������װ�䡢��ж�dz����㣬����õ��㷺��Ӧ�á�
2.1.2��������װ��
��ͼ2-1��ͼ2-2�У��г���һЩ���õı�ĥ��������������Щ��������������Դ��һ�㳣���л���ѧʵ��������Ҫ��ʵ��װ�ã����������������衢�������յ�װ�á�
Բ����ƿ ������ƿ ֱ��������
���������� ����������
��Һ©�� ��ѹ©��
ͼ2-1���ñ�ĥ������
����ͷ ��������ͷ
���β�ӹ� β�ӹ�
���� �¶ȼ���
ͼ2-2 ���ñ�ĥ������ ���������� ��ͷ
�ձ� ��ƿ
����©�� ��ĩ©�� ����©�� ����ƿ
ͼ2-3 ������������
ͼ2-4��һ�鳣������װ�á��������¶Ȳ�̫��ʱ������140�棩��ͨ��ѡ�����������ܣ���ͼ2-4��1����
ͼ2-4 ����װ�� ͼ2-5 ��Ӧװ��
��ֱ�������ܣ�����ǰ�߽�֮��������Ч��Ҫ��һЩ�����ʵ��Ҫ�������ˮ�����������϶˻�Ӧ���ø���ܣ���ͼ2-4��2�������������¶Ƚϸ�ʱ������140�棩����Ҫѡ�ÿ��������ܣ���ͼ2-1������Ϊ���λ�ֱ���������ڸ���������ը�ѡ�
ͼ2-5��һ�����õķ�Ӧװ�á�ͼ2-6��һ�鳣�õĽ��跴Ӧװ�á����ֻ��Ҫ����衢�����͵μ��Լ�������ͼ2-6��1����ʾװ�ü��ɡ��������Ҫ��������Ҫ���һ�Ҫ�������Է�Ӧ�¶ȣ������Ҫ����ͼ2-6��2���ͣ�3����ʾװ�ã�������Ŀ���ƿ��װ�䷴Ӧװ�á���Ȼ������ô��������������е�����������ö�����ƿ���ɡ��������ӵ��Լ��Կ�����ˮ���У���ӦҪ�������ˮ����Ӧ���ú�ѹ��Һ©������ܣ���ͼ2.6��3����ʾ����
��1�� ��2��
ͼ2-6���跴Ӧװ��ͼ
ͼ2-7��һ����������װ�á������Ӧ������������������Ǿ�Ҫ��װ��ͼ2-7��ʾ����������װ�á�
ͼ2.7 ��������װ��
��ͼ2-7���ձ�������ƿ�п�װ��һЩ��������Һ������Һ���Һ�� �����շ�Ӧ�����в����ļ��Ի��������塣
2.1.3ע������
��l��ĥ���������ճ����һ�𣬲���ʹ����ж�������õ紵�����ճ��ӿڴ����ȣ�Ȼ�������Ų�ж��
��2���Բ����������ȣ������Թ���һ�㶼����ֱ���û���ȣ��Է����ѡ�
��3����ڲ�������������ƿ���������ѣ��ʲ���ֱ�Ӷ�����ȡ���������������Ͳ���Ȼ�Ӱ�����ȷ�ȣ�ϴ���������ɶ��������ڸ����º濾��
��4�������������������Һ©��������ʱ��Ӧ�ý�������ĥ��֮����ֽƬ���뿪��������ճ�Ρ�
��5������õ�Һ©������������ƿ�Ȳ�������ʢװ������Һ��ʹ�����Ӧ��ʱϴ�ӣ��Է�ֹճ�ᡣ
��6����ϴ��������ʱ������ȥ�۷ۻ�����ϴ�ྫ����ϴ�ӣ���ϴ��ϣ�����ˮ�徻�������ڲ��������������ɡ������Ҫ�������ɣ�����������ͪ���Ҵ�������ϴ��ϴ�Ϻ��õ紵����
��7���ڻ���װ���У�һ���������������ܡ���Ϊ�����������ܽӴ�����ϴ�����Ч���Ϻã������ʺ��ڵͷе��ܼ��Ļ�����������������¶Ƚϸߣ�Ҳ�ɲ���ֱ�������ܡ���Ȼ���������¶ȸ���140��ʱ���ÿ��������ܡ�
��8���ڽ��跴Ӧ�У������Ӧ��������ϴ��ճ�������й������ʣ���ʱ���ô�������Ч�����ѣ���Ӧ�Ի�е����������Ϊ�ˡ�
��9���ڲ�����������װ��ʱ����ͼ2-7����Ӧ����ע��۲����������������ʱ����Ϊ��Ӧ�¶ȵı仯��������ϵ���γ�һ���ĸ�ѹ���Ӷ�������������Һ����������İ취�ܼ����ֲ���©�����������ڽ�������Һ��Һ���ϣ�ʹ��Ӧ��ϵ�������ͨ��������ѹ��
2.2����
Һ̬�������ȷ��ڻ�Ϊ������������������ת��ΪҺ�壬����������̾ͳ�������Distillation���������Ǵ����ͷ���Һ̬���ʵ�һ�ֳ��÷�����ͨ�������Բⶨ��Һ̬���ʵķе㡣
2.2.1ʵ��ԭ��
����Һ̬������һ��ѹ���¾���ȷ���ķе㣬��ͬ�����ʾ��в�ͬ�ķе㡣��������������ò�ͬ���ʵķе�����Һ̬�������з���ʹ�������Һ̬���������ʱ�����ڵͷе������ӷ������ȱ����������߷е��������ӷ���ӷ��������������ױ�����������������ƿ�У��Ӷ�ʹ�������Է��롣������ֻ�е���ַе������30������ʱ��������нϺõķ���Ч���������ַе���첻����Ҫ���÷��������Һ̬�������з���ʹ�����
��Ҫָ�����ǣ����к㶨�е��Һ�岢�Ƕ��Ǵ��������Ϊ��Щ�������֮������γɶ�Ԫ����Ԫ���л��������л�����Dz���ͨ������������з���ġ�ͨ������������ķг̣��е㷶Χ����С��Լ0.5��l�棩���������ķг̽ϴ���ˣ���������ȿ��������Եؼ��������Ҳ�������ж�������Ĵ��ȡ�
2.2.2ʵ�鷽��
��װ��������ƿ�������ܡ������ܺͽ���ƿ����ͼ2-8����Ȼ������Һ��ͨ��©����������ƿ���ڼ��뵽ƿ�У�Ͷ��l��2����ʯ���������¶ȼơ�
��ͨ����ˮ����ʼ���ȣ�ʹƿ��Һ����ڡ����ڻ��棬���������ٶȣ���1��2�Σ���Ϊ�ˡ�����������У�ע���¶ȼƶ����ı仯�����µ�һ�����Һ����ʱ���¶ȡ����¶ȼƶ����ȶ�������һ������ƿ�ռ���֡������Ȼ����ƽ�ȼ��ȣ�����������������������¶Ȼ�ͻȻ�½���������ö�����ѽ����꣬��ֹͣ���ȣ����¸ö���ֵķг̺������������������ֵ��¶ȷ�Χ��С���䴿�Ⱦ����ߡ�
��ʱ�����л���Ӧ��������Ҫ�Է�Ӧ�����ֱ������ʱ�����Խ�������ƿ������ƿ��װ������װ��ֱ�ӽ�������ͼ2-9����
ͼ2-8 ������װ��ͼ ͼ2-9 �ɷ�Ӧװ�ø�װ������װ��ͼ
2.2.3ע������
��1��������ƿ��С��ѡ����������Һ�����������ͨ����������Һ������Լռ������ƿ�����1/3��2/3��
��2����������Һ��ķе���140������ʱ��Ӧѡ��ֱ�������ܣ��е���140������ʱ����Ҫѡ�ÿ��������ܣ�������ֱ�����������������ѡ�
��3���������װ�������õĽ�������ܣ�������ܺͽ���ƿ֮��Ӧ���п�϶����ȷ������װ���������ͨ���������ϵ���Ⱥ�������¹ʡ�
��4����ʯ��һ�ִ�����Ե����ʣ����ش�Ƭ��ëϸ�ܡ���Һ�����ȷ���ʱ����ʯ�ڵ�С���ݾͳ�Ϊ�������ģ�ʹҺ�屣��ƽ�ȷ��ڡ���������Ѿ���ʼ��������Ͷ��ʯ����ʱǧ��Ҫֱ��Ͷ�ŷ�ʯ�������������С���ȷ�������ǣ���ֹͣ���ȣ���Һ������Ƭ�̺��ٲ��ӷ�ʯ��
��5������ͷе���ȼҺ�壨�����ѣ�ʱ��ǧ����������ȣ���ʱ������ˮԡ���ȡ�������е�ϸߵ�Һ��ʱ��������������ȡ��������ʱ����ƿ�ײ�һ��Ҫ�÷�ʯ�������Է������Ȳ��ȶ�ը�ѡ�
��6�����ۺ�ʱ������Ҫʹ������ƿ���ɣ��Է����⡣
2.3 ����
������ֻ�ܶԷе����ϴ�Ļ��������Ч�ķ��룬�����÷���������������ɶԷе�����Ļ������з�����ᴿ�����ֲ���������Ϊ����Fractional Distillation������˵��������Ƕ���������÷������������Խ��е����1��2��Ļ������뿪����
2.3.1ʵ��ԭ��
����������ȷ���ʱ�����������Ƚ������������������������²���������и߷е�����������������ȴ��������������������ƿ���Ӷ����¼��������������еͷе���ֵĺ���������ӡ���һ�����̿��Կ�����һ�μ������߷е�����Һ�ڻ���;��������������������ʱ������֮�䷢���Ƚ����������������У�ͬ���Ǹ߷е���ֱ��������ͷе���
�ּ������������ֿ��Կ�����һ�μ������������������ڷ������ڷ����ؽ����������������ͻ����Ĺ��̣�����˵���ظ��ؽ����Ŷ�μ�������ˣ�ֻҪ��������Ч���㹻�ߣ��ӷ������϶�������������־��ܽӽ��ͷе㵥��ֵĴ��ȣ����߷е�����Ի�����������ƿ�С���Ҫָ�����ǣ����ڹ��л������к㶨�ķе㣬������һ�����������Ҳ�����������빲�л���
2.3.2ʵ�鷽��
������������װ��Բ����ƿ����Ͷ�ż�����ʯ��Ȼ������װ���������¶ȼơ������ܡ������ܼ�����ƿ����ͼ2-10����
ͼ2-10 ����װ��ͼ
��ͨ����ˮ����ʼ���ȣ�ʹҺ��ƽ�ȷ��ڡ���������������ʱ��ע������¶ȣ�ʹ����ٶ�ά����2��3����һ�Ρ���¼��һ�����Һ�������ƿʱ���¶ȣ�Ȼ����ݾ���Ҫ��ֶ��ռ���֣�����¼����ֵķе㷶Χ�������
2.3.3ע������
��1������ƿ�������ܵ�ѡ��μ�2.1.2�е�ͼ2-1��
��2��������������Ӱ�����Ч�ʵ���Ҫ����֮һ��һ��������������Խ�ߣ���������������Һ����Ƚ���������Խ�࣬����Ч����Խ�á����ǣ�������������ߣ����Ӱ������ٶȡ�
��3���������ڵ������Ҳ��Ӱ�����Ч�ʵ�һ����Ҫ���ء���������������������������Һ�Ӵ������ã������ȱ����Խ��Խ��������߷���Ч�ʡ���������Ҫָ�����ǣ������֮��Ҫ����һ���Ŀ�϶������ᵼ���������ѡ�ʵ�����г��õ�Τ�ϣ�Vigreux����������һ�����ڳʴ�״�ļ��������������������ϡ�
��4�������½ϵͻ������Һ��ķе�ϸ�ʱ���������ľ������ܾͻ�Է���Ч�ʲ�������Ӱ�졣����������£�����������ľ������ܲ��ɢ�ȾͿ죬�������ά��������Һ��������ƽ�⣬�Ӷ�Ӱ�����Ч����Ϊ����߷������ľ������ܣ����ò������ȱ��²��Ͻ�������������
��5���ڷ�������У�Ҫע����ڼ����¶ȣ�ʹ����ٶ����С��������ٶ�̫�죬�ͻ����Һ����������Һ��������������ƿ�������ڷ��������γ�Һ������������������Ӧֹͣ���ȣ���Һ����ʧ�����¼��ȣ�ʹ��Һ�ﵽƽ�⣬�ٻָ��ռ���֡�
2.4 ˮ��������
��ˮ����ͨ�벻����ˮ���л����л�ʹ�л�����ˮ�������ж�����������������̳�Ϊˮ��������Steam Distillation����ˮ���������Ƿ�����ᴿҺ̬���̬�л����һ�ַ�����
2.4.1ʵ��ԭ��
���ݷ�ѹ���ɣ���ˮ���л����Ϲ���ʱ��������ѹΪ�����֮�͡���
P����� = Pˮ + P�л���
���ˮ������ѹ���л��������ѹ֮�͵��ڴ���ѹ�������ͻ���ڣ��л����ˮ�ͻ�һ����������Ȼ����������ʱ���¶�Ҫ����������һ��ֵķе㡣���仰˵���л�������ڵ�����е���¶������±��������������Ͻ������Һ���л��W�л����ˮ��Wˮ��������֮�ȣ�Ӧ�������ߵķ�ѹ��P�л����Pˮ������Է�������M�л����Mˮ���˻�֮�ȡ�
���磬��1-��������ˮ��������ʱ��l-������ˮ�Ļ������99.4����ڡ�ͨ�������ֲ�ѵ�֪����ˮ��99.4��ʱ������ѹ��99.18 kPa��744 mmHg��������ѹ���ɣ�ˮ������ѹ�� l-����������ѹ֮�͵��� 101.31 kPa��760 mmHg������ˣ�1-������ 99.4��ʱ������ѹ��Ϊ
2.13kPa��16 mmHg������ÿ���� 1gˮ���� 0.16 g 1-������������
�����л�����ˮ���ȷ��ڵ��¶�����100�����£���ˣ�ˮ������������ر��������ڸ���
�������仯���л�����롣��Ȼ���л��ﻹ���������Ϊ 0.7 kPa��5 mmHg��������ѹ���Ҳ�����ˮ�����⣬��Щ���д�����֬״���ʡ�ֱ����������ؽᾧ�ȷ������Է���Ļ����Ҳ���Բ���ˮ��������ķ��������롣
2.4.2ʵ�鷽��
����װˮ������������Բ����ƿ����������ͷ���¶ȼơ������ܡ������ܺͽ���ƿ���μ�ͼ2-11��1������������������ת����ƿ�У���T�ιܻ���������ˮ����������ʹˮ���ڡ�����ˮ������T�ι�֧�����ʱ����֧�ܿڹرգ�ʹˮ����ͨ����ƿ����ͨ��ȴˮ��ʹ�������������������Ѹ���������������ƿ������ٶ���2�Σ���Ϊ�ˣ�ͨ�����ڻ�����Կ��ơ������Һ�����������ٺ�����״��ʱ������ֹͣ�����ȴ�T�ι�֧�ڣ�Ȼ��ֹͣ���ȡ����ռ�Һת���Һ©�������÷ֲ㣬��ȥˮ�㣬���÷�����
�������ˮ����������������һ�ָ�Ϊ��ˮ��������װ��Ҳ���������ؽ���ˮ���������������ͼ2-11��2���������������Ҳ�ܼ��Ƚ��������л����������ˮ����Բ����ƿ�У���Ͷ�˼�����ʯ����ͨ����ˮ����ʼ���ȣ�����ƽ�ȷ��ڡ���������ͬǰ��������ͬ��ֻ�ǵ���ƿ�ڵ�ˮ���������ϵ����������ʱ����ͨ������ͷ�����õĵ�Һ©������ˮ�������װ��ͼ2-11��2������ˮ���������������ʹ����ヲ�������ܣ�ʹ���봿���ܵ�Ӱ�죬��ô����ͼ2-11��3���������Ϳ�����Ч�ر���������⡣���������ڿ�������ͷ��ܶνϳ���������������Ӱ����Ч����ʱ�������ò����Ⱦ��Ȳ��ϲ��ƣ��Ա�������Ѹ��ɢʧ���Ӷ��������Ч�ʡ�
ͼ2-11ˮ��������
2.4.3ע������
��1��ˮ������������һ��Ҫ���ð�ȫ�ܡ���ѡ��һ��������������ȫ�ܣ������¶�Ҫ�ӽ�ˮ�����������ײ���ʹ��ʱ��ע���ˮ��Ҫ���࣬һ�㲻Ҫ�������ݻ���2/3��
��2��ˮ��������������ƿ֮������ӹ�·Ӧ�����̣ܶ��Լ���ˮ�����ڵ�������е�����ġ�
��3������ˮ�����IJ�����Ӧ�����ӽ�Բ����ƿ�ײ��������������Ч�ʡ�
��4������������У�����н϶��ˮ������������������Բ����ƿ�У�������С�����ʯ������Բ����ƿ�ײ����ȡ�
��5��ʵ���У�Ӧ����ע��۲찲ȫ�ܡ�������е�ˮ�����ֲ�����������Ӧ������T�ιܣ�ֹͣ���ȣ��ҳ�ԭ���ų����Ϻ�����������
��6��ֹͣ����ʱ��һ��Ҫ�ȴ�T�ιܣ�Ȼ��ֹͣ���ȡ������ֹͣ���ȣ�ˮ��������������ȴ��������ѹ����ʹ��ƿ�ڵĻ��Һ����������
2.5 ��ѹ����
��Щ�л����������ȶ��Խϲ�����������¶Ȼ�δ������е���ѷ����ֽ⡢������ۺϡ����������Ĵ��������Ͳ��˲�ȡ��ѹ����ķ�������Ӧ���ڼ�ѹ�����½�������ѹ�����ֳ��������Vacuum Distillation�������Խ��л��������ڵ�����е���¶��������������ѹ���������ʺ���������Щ�е�ߡ����ȶ��Բ���л������
2.5.1ʵ��ԭ��
Һ�廯����ķе������ѹ�������еĹ�ϵ�������ѹ������ʱ��ʹҺ���������ݳ�����������Ҫ������Ҳ�ή�͡����仰˵������������ѹ����Һ��е�ͻ���֮�½������磬����ȩ�ڳ�ѹ�µķе�Ϊ 179�棯101.3 kPa��760 mmHg������ѹ������ 6.7 kPa��50 mmHg��ʱ����е��ѽ��͵� 95�档ͨ������ѹ�����͵� 2.67 kPa��20 mmHg��ʱ�������л�������ķе�Ҫ���䳣ѹ�µķе��100�����ҡ��е���ѹ���Ĺ�ϵ�ɽ��Ƶ���ͼ2-12�Ƴ������磬ijһ�������ڳ�ѹ�µķе�Ϊ 200�棬��Ҫ�� 4.0 kPa��30 mmHg���ļ�ѹ�����½��������������ô�������е��Ƕ����أ�������ͼ2-12�г�ѹ�е�̶������ҵ�200���ʾ�㣬��ϵͳѹ���������ҳ� 4.0 kPa��30 mmHg����ʾ�㣬Ȼ�����������ӳ�һֱ�߲����ѹ�е�̶����ӳ��ཻ���佻����ʾ�����־��Ǹû������� 4.0 kPa��30 mmHg����ѹ�����µķе㣬�� 100�档��û������������Դ������£��ɴ˷����ù���ֵ����ʵ�ʼ�ѹ����������Ǿ���һ���IJο���ֵ��
A����ѹ�е㣩 B����ѹ�е㣩 C��ϵͳѹ��mmHg��
ͼ2-12 Һ���ڳ�ѹ�ͼ�ѹ�µķе���ƹ�ϵͼ
2.5.2ʵ�鷽��
ͨ������ѹ����ϵͳ��������װ�á���ȫƿ����������װ�á�����ƿ����ѹװ����ɡ�������ѹ�������ʱ������װ��������ƿ����������ͷ�������ܡ���ս����ܼ�����ƿ���Բ���©��������������ע��������ƿ�У�����ëϸ�ܣ�ʹëϸ�ܾ����ӽ�ƿ�ף���ͼ2-13(1)����
����ս������ú�������Ƥ�������밲ȫƿ����ȴ�塢��ռơ�����������������ƿ���ͱ������ӣ���ͼ2-14������ȴ������ڹ�ڱ���ƿ�У���Һ�����-����ȴ����ȴ��
��1�� ��2��
��3��
ͼ2-13 ��ѹ����װ��
�ȴ�ȫƿ�ϵĻ�����ʹ��ϵ�������ͨ��Ȼ�����ͱó����������رհ�ȫƿ�ϵ�������ͬʱע��۲�ѹ���ƶ����ı仯��ͨ��С����ת��ȫƿ�ϵ�������ʹ��ϵ��նȵ���������ֵ��
��ͨ�������ϵ�����ˮ����ʼ����ԡҺ��������ƿ���ȣ�ͨ��ԡҺ�¶�Ҫ�߳����������ʼ�ѹʱ�ķе�30�����ҡ������ٶ���1��2�Σ���Ϊ�ˡ������������ʱ����¼��е㼰��Ӧ
��ѹ����������������������м��ֲ�ͬ�е����֣���ͨ����ת��ͷ�����ܡ��ռ���ͬ����֡�
ͼ2-14 ��ת������
���������ֹͣ���ȣ�������ȫƿ�ϵ���������ϵͳ�����ѹ���ﵽƽ��ر��ͱá�
��ʹ���ͱý��м�ѹ����ǰ��ͨ��Ҫ�Դ�����������Ԥ�����������ڳ�ѹ�½��м�����2.2����������ˮ�ü�ѹ��������ת����������ͼ2-14�����������ͷе���֡�
2.5.3ע������
��l���ڼ�ѹ����װ���У��ӿ�������ͷֱ������ƿ����ĩ����ϸ����ëϸ�ܣ����������������ĵ����ã�ʹ����ƽ�ȡ��������ƿ��װ����������ӣ��ڼ�ѹ��������У�����������������Ҳ�ɱ���ƽ�����������Ͳ��ذ�װëϸ�ܣ���ͼ2-13��2�����������������Կ������У��ڴ��������¼�ѹ����ͱȽϺ��ʡ���ʱ����ʹ��ëϸ�ܣ���Ӧͨ��ëϸ�ܵ���������壨�絪�����������Է�����
��2�����ͱú�Ҫע��۲�ѹ���ơ����������ϵѹ�����仯����ϵͳ���ܴﵽ�ͱ�Ӧ�ôﵽ����նȣ���ô�ü��ϵͳ�Ƿ�©�������ǰ�Ƚ��ͱùرգ��ٷֶβ���Щ���Ӳ�λ�����������װ��©��������������װ�õĸ������Ӳ�λ�ʵ���Ϳһ�����֬����ͨ����תʹĥ�ڽ�ͷ���Ǻ����ܡ�����������������ѹ���Ƶ������മ���ĽӺϲ�λ©������Ϳ�������ۻ���ʯ�������õ紵��������ڣ���Ϳ�����֬���������ϣ����ɰ�ʵ�鷽�������������ͱá�
��3����ѹ����ʱ��һ��Ҫ��ȡ��ԡ����ˮԡ���ķ������о��ȼ��ȡ�һ��ԡ��Ҫ�߳����������ڼ�ѹʱ�ķе�30�����ҡ�
��4��������������߷е����ʻ���۵����ʣ���ɲ���ͼ2-13��3����ʾװ�ý�������ʡȥ�����ܡ���������¶Ƚϸߣ��ڸ�������ʱ��Ϊ�˼���ɢ�ȣ����ڿ�������ͷ���ò����Ⱦ��Ȳ��ϲ�������������ڼ�ѹ�����£�Һ��е����140��150�棬������ˮԡ�Խ���ƿ��ȴ��
��5��ʹ���ͱ�ʱ��Ӧע������뱣��������ʹˮ�֡��л����ʻ���������������ڣ���������ؽ����ͱõ�Ч�ʡ�������װ�����ͱ�֮������װ�İ�ȫƿ����ȴ�塢����������������ƿ��Ŀ�ľ���Ϊ�˱����ͱá�����������ʱ��ͻȻ�������л���ϣ���ȫƿ���������á���ʱ������ϵͳ��ѹ������ͻȻ�仯���Ӷ����±��͵���������ƿ�����þͿ��Ա�����ͳ������������������⣬װ�ڰ�ȫƿ���ϵĴ�����˫ͨ�ܿ���������ϵͳѹ���������������Щ��������ķе�ϵ͵���֣����Ӿ����������ȴ����뵽ʢ��Һ����ɱ����-ˮ���-�ε���ȴ���Ĺ�ڱ���ƿ�н�����ȴ����������Ҳ�Ƹ�������һ����2��3������Щ�������зֱ�װ����ˮ�Ȼ��ơ�����״�������Ƽ�Ƭ״����ʯ������������ˮ�֡��������弰�������塣Ӧ��ָ�����ǣ������ͱü�ѹ����ǰ��һ��Ҫ�������������ˮ�ü�ѹ�����������ͷе����ʣ���ֹ�ͷе����ʳ����ͱá�
��6��ͼ2-15Ϊ���ʽˮ��ѹ���ƣ������ڲ�����ѹϵͳ����նȡ������۹���߶�֮�Ϊ��ѹϵͳ����նȡ�ʹ��ʱӦ��ע�⣬����ѹ��������ʱ��ҪС��������ȫƿ�ϵ�˫ͨ��������������������ϵͳ��ʹѹ�����е�ˮ����������ԭ���Ա�����ϵͳ�ڵ�ѹ��ͻ��ʹˮ�������Ʋ����ܡ�
ͼ2-15���ʽˮ��ѹ����
2.6 �۵�ⶨ
�ڴ���ѹ���£������������ɹ�̬ת��ΪҺ̬ʱ���¶ȳ�Ϊ�û�������۵㡣�۵��ǹ����л����������������֮һ��ͨ���ⶨ�۵㲻�����Լ���ͬ���л���������һ����ж��䴿�ȡ�
2.6.1ʵ��ԭ��
�ϸ��˵����ν�۵�ָ�����ڴ���ѹ���»�����Ĺ�-Һ����ﵽƽ��ʱ���¶ȡ�ͨ�������л������ﶼ����ȷ�����۵㣬���Ҵӹ�����۵�ȫ�۵��¶ȷ�Χ�����۳̻��۾ࣩ��խ��һ�㲻����0.5��1�档���ǣ������Ʒ�к������ʣ��ͻᵼ���۵��½����۾�������ˣ�ͨ���ⶨ�۵㣬�۲��۾࣬���Ժܷ���ؼ���δ֪����ж��䴿�ȡ���Ȼ����һ���ʿ������������־����������ͬ�۵�Ļ�������Ƿ�Ϊͬһ���������ʮ�ּ�ֻҪ�������ֻ���������һ�𣬲��۲����۵㡣����۵��½��������۾������DZض����������ʲ�ͬ�Ļ������Ҫָ�����ǣ����������������ʱ�����ֽ⡣��ˣ���ʹ�䴿�Ⱥܸߣ�Ҳ������ȷ�����۵㣬�����۾�Ͽ���
2.6.2ʵ�鷽��
��������Ĵ�����Ʒ�÷��ڸ���ྻ�ı������ϣ��ò�����������ϸ��Ȼ���ò��۵�ëϸ�ܿ��ڵ�һ�˴�ֱ�����ĩ״����Ʒ�У�������������Ʒ����ëϸ�ܡ��ٽ�ëϸ�ܿ��ڶ˳��ϣ���ëϸ�ܷ�ڶ���ʵ��̨��������£���Ʒ������ëϸ�ܵײ�����˲����������Σ�Ȼ����ëϸ�ܷ�ڶ˳��£���һ��Լ50cmֱ���ڱ������ϵIJ��������������£������������Σ�ʹëϸ���е���Ʒװ�����ܾ��ȣ���Ʒ��Լ 4 mm��Ȼ��װ����Ʒ��ëϸ����ϸ��ƤȦ�̶����¶ȼ��ϣ���ʹëϸ��װ����λλ��ˮ������ͼ2-16����
ͼ2-16 �۵�ⶨװ��
�����գ�Thiele���۵�ⶨ�̶ܹ�������̨�ϣ�ע�뵼��Һ��ʹ����ҺҺ��λ�������۵�ⶨ�ܽ���ڴ����ܿ����ÿ���С�۵��������������в���ëϸ�ܵ��¶ȼƲ������У�ʹ�¶ȼƵ�ˮ����λ�������۵�ⶨ����֧�ܵ��м䡣
�ֲ�ʱ����С���������۵�ⶨ�ܵײ����ȣ������ٶ���5�棯minΪ�ˡ���ϸ�۲��¶ȵı仯����Ʒ�Ƿ��ۻ�����¼��Ʒ�ۻ�ʱ���¶ȣ����������Ĵֲ��۵㡣��ȥ���棬�õ���Һ�¶Ƚ����ֲ��۵�����Լ30�棬���ɲο��ֲ��۵���о��⡣
����ʱ�����¶ȼƴ������۵�ⶨ����ȡ�������ϵڶ����۵�ܺ��ɼ��Ȳⶨ����ʼ���¿��Կ�һЩ��Լ5�棯min�����¶�������ֲ��۵�Լ10��ʱ��Ҫ���������ٶ���1�棯min���ҡ�����۵���е���Ʒ�������䡢ʪ���������ֳ�СҺ�Σ���������ʼ�ۻ�����¼��ʱ���¶ȣ��������¶ȣ����������������£�ֱ����Ʒȫ�ۣ���¼ȫ�ۣ������о��ֹ������ۻ���ֻʣ����������ʧ��ϸ�پ��壩ʱ���¶ȡ������ۻ����̲μ�ͼ2-17��
��Ʒ��ʼ̬ �������� �ճ���Һ�� ������ʧ�ľ��� Һ��
ͼ2-17 �����ۻ�����ʾ��ͼ
2.6.3ע������
��1���������۵�ⶨ�ܲⶨ�۵���ʵ�����г��õ�һ�ֲⶨ�۵�ķ��������⣬���ɲ������۵�ⶨ�ǻ������۵��ǡ����У������۵�ⶨ�Dzⶨ�۵����ʹ����Ʒ�١��ɲ���۵���Ʒ���ɹ۲���Ʒ�����ȹ����еı仯���ص㡣
��2��������Ʒһ��Ҫ����ָ�����ٽ��вⶨ�۵㡣������ˮ�ֵ���Ʒ�ᵼ�����۵��½����۾��������⣬��Ʒ��Ӧ�����ϸ��װ��Ҫ���ܾ��ȣ�������Ʒ�����䴫�Ȳ��ȣ�Ҳ��ʹ�۾�����
��3�����Ƚ��ʵ�ѡ��ɸ��ݴ������ʵ��۵���������۵���95�����£�������ˮ������Һ�����۵���95��220�淶Χ�ڣ���ѡ��Һ��ʯ���ͣ����۵��¶��ٸ�Щ������Ũ���ᣨ250��270�棩������ע�ⰲȫ��
��4�����������۵�ⶨ��ע�뵼��Һʱ��Ҫ������Ҫ���ǵ�����Һ���Ⱥ�����������͵����ء����⣬���ڹ̶��۵�ܵ�ϸ��ƤȦ��Ҫ���뵼��Һ�У������������䡣
��5����Ʒ���ⶨ�۵���ȴ���ֻ�ת��Ϊ��̬�����ڽᾧ������ͬ���������ͬ�ľ��͡�ͬһ������IJ�ͬ���ͣ����ǵ��۵㳣����һ������ˣ�ÿ�β��۵㶼Ӧ��ʹ����װ��Ʒ���۵�ܡ�
2.7�е�ⶨ
������Һ����������������ѹ�봦��ѹ�����ʱ�ͻ���ڣ���ʱ���¶Ⱦ��Ǹ����ʵķе㣨 Boiling Point�����bp�����е����л����������������֮һ��ͨ���ⶨ�е���Լ����л���������ж��䴿�ȡ�
2.7.1ʵ��ԭ��
��һ��ѹ���£�ÿһ�ֻ����ﶼ�����ض��ķе㡣���仰˵��ͬһ�ֻ������ڲ�ͬ��ѹ���£���е��Dz�ͬ�ġ���ˣ�����һ�ֻ�����ķе㳣Ҫע����ѹ�����������磬������ͪ��13.3 kPa��100 mmHg��ʱ�������¶�Ϊ 224.4�棬��Ϊ 224.4�棯13.3 kPa�����������ָ�����䳣ѹ�µķе㣬��ͨ����ע��ѹ�����������磬������ͪ�ķе��Ϊ305.4�棬ָ�ľ��dz�ѹ�µķе㡣��Ҫָ�����ǣ����к㶨�е��Һ�岢��һ�����Ǵ��������Ϊ���л����Ҳ���к㶨�ķе㡣��ˣ��ⶨ�е�ֻ�ܶ��Եؼ���һ���������������һ����֪����ԣ�ͨ������е㣬����г̷�Χ�ǿ����ж��䴿�ȵġ���Ϊ����������ķг�һ���խ��ԼΪ0.5��1�档�ⶨ�е��г����������������������õ�������װ�ã��䷽��������������ͬ����2.2�ڣ�����������ʹ�õ�װ�����۵�ⶨװ�����ơ�
2.7.2ʵ�鷽��
���ھ�3��4mm����8��10cm��һ�˷�ڵIJ��������е�ܣ�����ڵμ�1�δ���Һ�塣����һ���ھ�Լ lmm����Լ 9 cm�IJ���ëϸ�����ڹܣ��ڹ�һ���Ƿ�յġ����ڹܿ��ڶ����²���е���У���ͼ2-18������С��ƤȦ���е�̶ܹ����¶ȼ��ԣ�ʹ�е�ܵ�λ���¶ȼ�ˮ����λ�������������۵�ⶨ���У���ͼ2-16�����������ȣ��������£����û�۲쵽�����ݴӷе���ڵ�Һ�����ݳ������������ڹ��е����������������¡���������Һ��ķе�ʱ���е���н���һ���������ݿ����ݳ�����ʱ������ֹͣ���ȣ���ԡҺ������ȴ�����������ݳ����ٶȽ�������������һ��������Һ���ӿ��������ڹ���ʱ���ڹ��ڵ�����ѹ�����ѹ��������ȣ���ʱ���¶ȼ�Ϊ��Һ���ڳ�ѹ�µķе㡣
ͼ2-18 �����е�ⶨ��
2.7.3ע������
��1���ⶨ�е�ʱ�����Ȳ�Ӧ���ͣ��������ڽӽ���Ʒ�ķе�ʱ�����¸�Ҫ��һЩ������е���ڵ�Һ���Ѹ�ٻӷ����������ⶨ��
��2������ڼ��Ȳⶨ�е�����У�û�ܹ۲쵽һ����С���ݿ����ݳ��������Ƿе��ڹܷ�ڴ�û���֮�ʡ���ʱ��Ӧֹͣ���ȣ���һ���ڹܣ�������Һ�¶Ƚ���20������²ⶨ��
2.8 �ؽᾧ
���ñ�����������������ͬһ�ܼ��е��ܽ����ܵIJ��죬�������IJ�����Ϊ�ؽᾧ��Recrystallization�����ؽᾧ�Ǵ��������л���������õ�һ�ַ�����
2.8.1ʵ��ԭ��
�����л������ܼ��е��ܽ�����¶ȵ�Ӱ��ܴ�һ����˵�������¶Ȼ�ʹ�ܽ�����������¶���ʹ�ܽ�ȼ�С������������л����Ƴ��ȵı�����Һ��Ȼ��ʹ����ȴ����ʱ�������ܽ���½���ԭ���ȵı�����Һ�ͱ������Ĺ�������Һ������о�����������ͬһ���ܼ����ԣ����ڲ�ͬ�Ĺ��廯������ܽ����Dz�ͬ�ġ��ؽᾧ�����������ò�ͬ�������ܼ��еIJ�ͬ�ܽ�ȣ����߾��ȹ��˽��ܽ��Բ�������˳����������ܽ��Ժõ���������ȴ�ᾧ�����Ա�����ĸҺ�У��Ӷ��ﵽ���봿����Ŀ�ġ�
2.8.2ʵ�鷽��
��l�������ؽᾧ��
����1g���ϵĹ�����Ʒ������һ�㶼���ó����ؽᾧ�������Ƚ����ؽᾧ���л���װ��Բ����ƿ�У��������ڹ��������ܼ���Ͷ�뼸����ʯ�����û��������ܣ���ͼ2-4������ͨ����ˮ���������У�����ʱ��ҡ����������в��ֹ���û���ܽ⣬����������ܼ��������ֻ���������ܼ��ķе�ϵͣ�������ȫ���ܽ��������һЩ�ܼ�������ԼΪ�Ѽ����ܼ�����15����
�����Һ�к���ɫ���ʣ����Բ��û���̿��ɫ���������֮̿ǰ��һ��Ҫ��������Һ����ȴ���Է����𱩷С��������̿����һ��Ϊ���ؽᾧ�л���Ͷ������1����5�����������ȣ�����5��10 min���þ�Ԥ�ȹ��IJ���©�����ȹ��ˣ��˳����������ʺͻ���̿��������Һ������Ȼ��ȴ�����£�ʹ����������Ȼ���������¹��ˣ��Գ�ȥ���ܼ����ܽ�ȴ�ġ��Բ�����ĸҺ�е����ʡ��˳�ĸҺ�����������ܼ��Թ����ռ���ϴ�Ӽ��Σ���ɺ����÷��ڱ������Ͻ��и������Ĵ��ȿɲ����۵�ⶨ�����г���������
��2�������ؽᾧ��
�����������Ʒ����ʱ������ 500 mg��������ͨ����©�����ؽᾧ�����DZȽ����ѵģ�һ����ʧ�ϴ���Y��ɰо©����������ʮ�ַ��㣬������ʧ��С����ͼ2-19����
����ʱ���Ƚ���Ʒ�ɲ����ܿڷ������У����������ܼ������ڲ����ܵ��ڵ���Ʒ��ϴ��ȥ���ò���������ԡ����ˮԡ�м������У����õι������в����ܼ���ֱ����Ʒȫ���ܽ⡣ֹͣ��ԡ���������������ϵ��ͼ���ˮ����Ȼ��Ѹ�ٽ��������ã�����Ƥ����ͨ����������Y��©���ڼ�ѹ��ʹ©�����ȵı�����Һ����ɰо©�����뾻��������У����á��ᾧ��
ͼ2-19 �����ؽᾧ
2.8.3ע������
��l��ѡ���ʵ����ܼ����ؽᾧ������һ����Ҫ�Ļ��ڡ���ѡ�ܼ�Ӧ�þ߱�����������������������ʷ�����ѧ��Ӧ�����������ʺ���������ѡ�ܼ��е��ܽ�������ԵIJ��죬�����Ǵ������������ܼ��е��ܽ��Ӧ���¶ȵı仯�������IJ��죻���⣬�ܼ�Ӧ�������ؽᾧ���ʷ��롣�����ѡ�ܼ����������������������Ҿ��á���ȫ������С�����գ��Ǿ������ˡ�
��2�������ѡ�ܼ���ˮ������Բ��û���װ�á���ʹ���ӷ����л��ܼ���һ�㶼Ҫ���û���װ�á�
��3���ڲ����ӷ��ܼ�ʱͨ��Ҫ����������ܼ����������ȹ��˲����У����ܼ�Ѹ�ٻӷ����¾����ڹ���©�������������⣬��������ȼ�ܼ�ʱӦ��ע��ܿ�����
��4����Һ��������ɫ���ʣ���ʹ�����ľ�����Ⱦ��������֬״���ʸ���Ӱ���ؽᾧ������������������������û���̿��������ͨ��������̿�ڼ�����Һ����ˮ��Һ���е���ɫЧ���Ϻã����ڷǼ�����Һ�е���ɫЧ��Ҫ��һЩ����Ҫָ�����ǣ�����̿���������ʵ�ͬʱ���Դ���������Ҳͬ�������������á���ˣ�����������ɫ��ǰ���£�����̿������Ӧ�����١�
��5���ȹ��˲������ؽᾧ�����е���һ����Ҫ�IJ��衣�ȹ���ǰ��Ӧ��©�����ȳ��Ԥ�ȡ��ȹ���ʱ����ҪѸ�٣��Է�ֹ�����¶��½�ʹ������©����������
��6���ȹ��˺�������ҺӦ���侲����ȴ�ᾧ�������Һ���ѳ�����״�ᾧ�������ʵ�����ʹ���ܽ⣬Ȼ����Ȼ��ȴ���������Ի�ýϺõĽᾧ��
��7������ȴ���ᾧ�����˺����õ�ĸҺ���������¾���һ��ʱ�䣬��������һЩ���壬���䴿�ȾͲ����һ�����塣������ڽᾧ������һ����Ҫ��ǰ�������ᾧ�Ͳ��ɻ����һ��
��8������Y�ι��ȹ���ǰ��һ��Ҫ����Ʒ��Һ�IJ��������������ڵ��ù���ʱ�������ڲ�������Һ���ܻ���Ⱦ��Һ��
2.9��ȡ
���ܼ��ӹ����Һ����������ȡ����Ҫ�����ʣ���һ�������̾ͳ�Ϊ��ȡ��Extraction������ȡ��������ȡ�ʹ����л��������һ�ֳ��÷��������ҿ�������ϴȥ������е��������ʡ�
2.9.1ʵ��ԭ��
��ȡ������ͬһ�����������ֻ������ܵ��ܼ��о��в�ͬ�ܽ�ȵ����ʣ������һ���ܼ�ת�Ƶ���һ���ܼ����Ӷ��ﵽ������ᴿĿ�ĵ�һ�ַ�����
��һ���¶��£�ͬһ�����ʣ�M�������ֻ������ܵ��ܼ���A��B������ѭ���·���ԭ����
K=(WM/VA)/(W,M/V,B)
ʽ�У� K��ʾ���䳣���� WM/VA��ʾ M��������Ϊ V���ܼ���A�������ܽ�Ŀ�����W����W,M/V,B��ʾ M��������Ϊ V�����ܼ���B�������ܽ�Ŀ�����W������
���仰˵�����ʣ�M�������ֻ������ܵ��ܼ��е��ܽ��֮�ȣ���һ���¶�����һ����������ʽҲ���Ը�дΪ��
K=(WM/W,M)��(V,B/VA)
�ɼ����������ܼ���������ʱ�����䳣��K�͵������ʣ�M�����������ܼ��е��ܽ��֮�ȡ���Ȼ����������ܼ���������ܽ������е����ʣ�M����Ҳ�����ӡ�
�����Ϲ�ʽ�������Ƴ�������һ�������ܼ�������ȡ���ִ���ȡ��һ����ȡ��Ч�ʸߡ���Ȼ���Ⲣ����˵��ȡ����Խ�࣬Ч�ʾ�Խ�ߣ�һ������ȡ����Ϊ�ˣ�ÿ��������ȡ��Լ�൱�ڱ���ȡ��Һ�����1/3��
���⣬��ȡЧ�ʻ����ܼ���ѡ��������ء�һ��������ѡ���ܼ��Ļ���ԭ���ǣ��Ա���ȡ�����ܽ�Ƚϴ���ԭ�ܼ�������ܣ��е�͡�����С�����磬��ˮ����ȡ�л���ʱ�����ȷ¡�ʯ���ѡ����ѡ������������ܼ��������л�����ϴ�����е����������ˮ��������ʱ���ɷֱ���ϡ���ϡ���ֱ����ˮϴ�ӡ�
�������������Һ-Һ��ȡ���ԡ����Ҫ�ӹ�������ȡijЩ��֣�����������Ʒ�б���ȡ��ֺ�������ͬһ�ܼ��о��в�ͬ�ܽ�ȵ����ʽ�����ȡ�ͷ���ġ���ʵ�����У�ͨ�������ϣ�Soxhlet����ȡ����Ҳ��֬����ȡ�����ӹ�������������ȡ�������乤��ԭ����ͨ�����ܼ����Ȼ��������ú�������ʹ���������������ܼ�����ȡ��
2.9.2ʵ�鷽��
(1)Һ-Һ��ȡ��
����Һ©������̶�������̨�ϵ���Ȧ�У��Ѵ���ȡ���Һ�����ΪV������ȡ�������ԼΪV��3�������Һ©�����Ǻ��Ͽ�������������ס��Һ©���Ͽڣ���������ʳָ��ס�Ͽ�����������ס��Һ©���¶˵Ļ�����λ��С����ʹ��ȡ���ʹ���ȡ���Һ��ֽӴ��������У�Ҫ��ʱ��©��β��������б�����������ų��������������壨��ͼ2-20���������������ظ����κ���Һ©�����÷�����Ȧ�ϣ����÷ֲ㡣�����������ȴ�Һ©���Ͽ�����Ȼ�������ʹ�²�Һ�������״�©���¿������ų����ϲ�Һ��©���Ͽڵ�������������ȡ������ű���ȡ���ʴ�ԭ������з��������һ����������ȡ���ξͿ����ˡ�����ȡҺ�ϲ����������ͨ������������ȡ���Ϳ��Ի����ȡ�
ͼ2-20 ��ȡ����ʾ��ͼ
��2��Һ-����ȡ��
������ȡ����ϸ������ֽ��������ϸ�����Σ���Բ��״��������ȡ���ڡ���Բ����ƿ�����ܼ�����Ͷ�ż�����ʯ�����������ܣ���ͼ2-21������ʼ���ȣ�ʹ�ܼ����ڣ����ֻ�������Һ���ϵ�����ȡ���У��ܼ����ۡ�����Һ��߳������ܶ���ʱ��������Ʒ����ȡҺ����Զ�������ƿ�С��ܼ����Ⱥ��ֻᱻ�������ܼ������������ֻ�������ȡ�ܣ���˷�����ʹ��ȡ�ﲻ�ϵػ�������ƿ�С�����ȡ������ϱ���ȡ�����������ܼ������ɻ����ȡ�
2.9.3ע������
��1�����÷�Һ©�����ݻ�һ��Ҫ�ȴ�������Һ�������1��2�����ڷ�Һ©���Ļ�����ӦͿ�ϱ���һ�㷲ʿ�֣�ע�ⲻҪĨ�ڻ������С�Ȼ��ת������ʹ�������������ȡ����֮ǰ��Ӧ�ȼ���������ˮ�Լ��������Ƿ��©��
ͼ2-21 Һ-����ȡ
��2����ʹ�õͷе��ܼ��������ѣ�����ȡ��ʱ����ʹ��̼������Һϴ�Ӻ���Һ��ʱ��Ӧע����ҡ��������Ҫ��ʱ�ط���������Һ©���е�Һ���״��Ͽ����������
��3������������У�Һ������黯������ͨ������ǿ����ʣ���ʳ�Σ����顣
��4����Һʱ�����һʱ��֪��һ������ȡ�㣬�����ͨ���ټ���������ȡ�����жϣ����������ȡ��������Һ©���е��ϲ�Һ�����²�Һ�����²�����ȡ�ࣻ��֮�����ϲ�����ȡ�ࡣΪ�˱������ʧ����ý���������Һ�嶼����������������
��5���ڷ�Һʱ���ϲ�ҺӦ��©���Ͽڵ�����������ȡ������Ⱦ��
��6���������ȴ����Һ��ӷ�Һ©���¶�����������Ӧ���©���Ͽ����Ƿ������Ͽ����Ѵ�Һ����Ȼ�Ų������Ǿü��������Ƿ�����
��7����������ȡ������ȡ���ʣ����������ŵ��ǽ�ʡ�ܼ������������ڱ���ȡ��Ҫ����ƿ�г�ʱ�����ȣ����������ֽ���ױ�ɫ�����ʾͲ��˲������ַ��������⣬Ӧ��������ȡ������ȡ����ʹ�õ��ܼ��ķе�Ҳ���˹��ߡ�
2.10����
�����������Ⱥ����ھ�ֱ��ת��Ϊ��������������������ֱ��ת��Ϊ���壬������̳�Ϊ������Sublimation���������Ǵ��������л����һ�ַ��������������������Է�����в�ͬ�ӷ��ȵĹ���������һ��ܳ�ȥ�ѻӷ������ʡ�һ���������ᴿ�õ��Ĺ����л��﴿�ȶ��ϸߡ����ǣ����ڸò����Ϸ�ʱ��������ʧҲ�ϴ������������ͨ��ֻ����ʵ�����������ʵľ��ơ�
2.10.1ʵ��ԭ��
�����˵���������ɹ�������ֱ�ӻӷ���������Һ������������������������ֻҪ�Dz�����Һ̬��ֱ��ת��Ϊ���壬��һ���̶���Ϊ������һ����˵���ܹ�ͨ�������������д�������������Щ���۵��¶����¾��нϸ�����ѹ�Ĺ������ʡ��������ʾ�������㣬���̡�Һ�������ಢ��֮�㡣һ�����ʵ��۵㣬ͨ��ָ���Ǹ����ʵĹ̡�Һ�����ڴ���ѹ�´ﵽƽ��ʱ���¶ȡ���ij���ʵ������ָ���Ǹ������ڹ̡�Һ��������ﵽƽ��ʱ���¶Ⱥ�ѹ��������������£�����ֻ�й̡������ࡣ��ʱ��ֻҪ���¶Ƚ��͵���������£������Ϳɲ���Һֱ̬��ת��Ϊ��̬����֮�������¶����ߣ����̬�ֻ�ֱ��ת��Ϊ��̬���ɴ˿ɼ�����������Ӧ����������¶����½��С����磬���������������¶��� 186�棬ѹ��Ϊ 104.0 kPa��780 mmHg������������185��ʱ���������Ѵ� 101.3 kPa��760 mmHg�����������鼴���ɹ��ೣѹ��ֱ�ӻӷ�Ϊ������
���⣬��Щ�����������ʱ��ƽ������ѹ�Ƚϵͣ��ڳ�ѹ�½�������ʱЧ���ϲ��ʱ���ڼ�ѹ�����½�������������
2.10.2ʵ�鷽��
��������������ϸ���÷����������У�Ȼ����һ����������С����ֽ��������������ϣ�����һ����©����������ֽ���棬��©���ľ�������һ�����ɵ������μ�ͼ2-22������С�����ʯ�����������ȣ�ʹ�������е�����������������������ֽС�������������ڲ���©���ı��ϣ���ֽ����Ҳ��ᾧ��һ���ֹ��塣������ϣ����ò���ֹγ�������©�������Լ���ֽ�ϵĽᾧС�Ĺ��䲢�ռ�������
��ѹ�����µ�����������������ѹ��������������ͬ�����Ƚ������������÷������˹��ڣ�
Ȼ�������˹�������ָ�������ܣ���ͨ����ˮ������ԡ���ȣ����˹�֧�ڽ�ˮ�û��ͱã��μ�ͼ2-22����
2.10.3ע������
��1������������Ҫ����ָ����������������ʱ�����л������ˮ����һ��ӷ�������Ӱ�����Ч����
��2�����������ϸ���һ�㲼��С����ֽ����Ҫ��Ϊ�����������Ϸ��γ�һ�²�㣬ʹ�ݳ����������������ڲ���©�����ϣ�����������������ʡ���Ҫʱ�����ڲ���©������Ϸ������²�������������
��3��Ϊ�˴ﵽ���õ���������Ч������ò�ȡɰԡ����ԡ������������ֱ�Ӽ��ȣ�ʹ�����¶ȿ����ڴ��������ʵ�������¶����¡���������¶ȸ���������¶Ⱦͻ�ʹ��ͬ�ӷ��Ե�����һͬ�������Ӷ����ͷ���Ч����
ͼ2-22 ����װ��
2.11ɫ��
ɫ����Chromatography��Ҳ��ɫ�㷨����������Ƿ��롢�ᴿ�ͼ����л����������Ҫ����֮һ��ɫ�����Դ�ڶ���ɫ���ʵķ��룬������������������Ÿ�����ɫ���������������룬��Ӧ�÷�Χ������չ����ɫ���ʡ�
2.11.1ʵ��ԭ��
ɫ�����������࣬������ԭ����һ�µģ������ô����������еĸ������ijһ�����У������ʳ����̶��ࣩ�����Բ��죬�������Բ��졢�ܽ��ԣ���Ʒ������ã�����ȣ��û������Һ��������������ࣩ�����̶��࣬ʹ�������������̶���֮����з����������������ã��Ӷ�ʹ������еĸ���ֵ��Է��롣���ݲ�ͬ�IJ���������ɫ���ɷ�Ϊ��ɫ�ף�Colum Chromatography����ֽɫ�ף�Paper Chromatography��������ɫ�ף�Thin Layer Chromatography����� TLC��������ɫ�ף�Gas Chromatography����
2.11.2ʵ�鷽��
��1����ɫ��
ѡһ���ʲ�������ϴ�������ֱ�̶�������̨�ϣ��������¶���һ����ƿ����ƿ���μ�ͼ2-23��������������¶�û��ɰо�������ӦȡһС����֬�����ޣ��ò����������������ף�Ȼ��������һ��Լ 1cm���ɰ���رղ����˵Ļ����������ڵ����ܼ������ߵ��ķ�֮������Ȼ��һ����������������֧�ּ������ܼ����ɺ�״��������Ӳ������϶�������һ��һ�����ӣ�ͬʱ�������¶˵Ļ�����ʹ�ܼ�����������ƿ���������������Ĺ����У�����ľ���Թܼл�������Ƥ�ܵIJ��������������������
ͼ2-23 ��ɫ��
��ʹ���������ȳ�����������ϣ������������渲��Լ 1cm���ɰ�㡣�������ӹ����У�Ӧ�����ܼ�Һ��ʼ�ո߳����������棨��ͼ2-23����
�����ڵ��ܼ�Һ�潵������������ʱ���رղ������¶˵Ļ������õιܽ��������õ���Ʒ��Һ�μӵ��������������㡣�õι�ȡ�����ܼ�ϴ�Ӳ������ڱ���մ�е���Ʒ��Һ��Ȼ�������ʹ�ܼ���������������ҺҺ�潵������������ʱ����ɼ���ϴ�Ѽ�����ϴ�ѡ������������������ɫ�����Ը��ݲ������г��ֵ�ɫ���ռ�ϴ��Һ������������ɫ�������ȷ��ռ����ռ���Ȼ���ñ���ɫ����һ�������ٽ���ͬ��ֵ��ռ�Һ�ϲ���һ�������ܼ������ø���֡�
��2������ɫ��
�� 5 g�轺G�ڽ������������뵽 12 mL l�����ȼ���ά���ƣ�CMC��ˮ��Һ�У����ɺ�״��Ȼ��״��Һ���ڽྻ���ز�Ƭ�ϣ�����������ʹͿ�����ƽ������Լ���� 8 �� 3 cm�ز�Ƭ6��8�顣���������ɣ�Ȼ����110������ڻ0.5 h��
�õͷе��ܼ��������ѡ���ͪ���ȷµȣ�����Ʒ���l�����ҵ���Һ��Ȼ�����ھ�С�� 1mm��ëϸ�ܵ���������ǰ������Ǧ���ڲ������Ͼ�ĩ�� 1cm�����ửһ���ߣ�Ȼ����ëϸ����ȡ��Һ�ں�����������������Ҫ���µ�����һ��Ҫ��ǰһ�ε���������ܼ��ӷ����ٵ�������������ߵ����һ��ߵ�ֱ�������� 2 mm�������ͬһ�鱡����ϵ������������ߵ���Ӧ���� 1��1.5 cmΪ�ˡ������Ϳ��Խ��в���չ����
�Բ�������չ����������չ������������Һ��߶� 0.5 cmΪ�ˡ���չ�����п�ƿ�ڷ���һ����ֽ��ʹ���������ڴﵽ��Һƽ�⡣��ֽȫ�����ܼ���ʪ��������ı�չ��б�������У�ʹ����һ�˳��£����ֵ����ߵ���չ����Һ��֮�ϣ����ϸ��ӣ���ͼ2-24������չ�����������뱡չ���϶�Լ 1cm��ʱ������չ��ȡ��������Ǧ�ʱ��չ������ǰ��λ�á�������
������ɹ۲�ߵ��λ�á�����ߵ�����ɫ���ɽ�������÷���װ�м����⾧�Ĺ��ƿ�ڸ���ƿ�ǡ���������ϳ������Եİ���ɫ�ߵ���ɽ���ȡ������������Ǧ�ʱ���ߵ��λ�á�Ȼ�������ߵ�� Rfֵ��
ͼ2-24 ֽɫ��װ��
��3������ɫ��
ѡ��һ������ྻ�ҳ������˵IJ���ֹܣ���ʱҲ���ò����ܣ��������������������ݻ���ȡ�ȸ��ݻ��Զ�һ��ĵ��壬����ȡ�൱�ڵ�������5����25���Ĺ̶�Һ���ú͵������൱�ĵͷе��ܼ������һ�𣬽�����ȡ�Ȼ��������ת�����ǣ����ú���Ƽ��ȣ������ܼ�����Ϳ�й̶�Һ�ĵ������� 110��120��ĺ������ϻ�2 h��
��ѡ�õIJ�����һ���Բ���ë��ס��������ձ���������һ������һ��С©����������ձã����ϻ����ĵ�������©���У�ʹ�����������ڡ���װ�������У�Ӧ�����û���ɫ������ʹ������������װ�þ������ܡ�װ�ϣ���©����ȥ���ò���ë��ɫ�����˶˶�ס�����Դ˶���Ϊ��������ɫ������������ͼ2-25����
ͼ2-25 ����ɫ��
ɫ������װ��ɫ���ǵ������У�Ȼ������������������������Լ 10��5 mL��min���Ͳ����¶ȣ��Ը���ʵ��Ҫ����¶ȣ������ڹ̶�Һ���ʹ���¶ȣ�������¼�ǻ���ƽ�Ⱥɽ����ⶨ��
2.11.3ע������
��1������ɫ����������Ӧ�ÿ��ǵ������������ʡ��ܼ��ļ��ԡ����ӵĴ�С�ߴ硢���������������Լ�ϴ�ѵ��ٶȵ����ء�
��2����������ѡ��һ��Ҫ���ݴ�����Ļ���������Ͷ��������������������ʺ��ڷ���
�����������Ի���������������ʺ��ڷ��밷������������������ڷ������Ի���
��轺�����ܱȽ��ºͣ������ζ�����ʣ��Ծ����ԣ��ʺ��ڼ��Խϴ�����ʷ��롣��
�紼�����ᡢ����ͪ�����ȡ�
��3���ܼ���ѡ��һ����ݴ����뻯����ļ��ԡ��ܽ�ȵ����ض�������ʱ��ʹ��һ�ֵ�
���ܼ�����ʹ������и���ַ��뿪������ʱ������Ҫ���û���ܼ�����ʱ����ʹ�ò�ͬ��
�ܼ�����ϴ�ѡ����磬�Ȳ���һ�ַǼ����ܼ��������������еķǼ�����ִ�����ϴ�ѳ�
����Ȼ����ѡ�ü����ܼ���ϴ�Ѿ��м��Ե���֡����õ��ܼ��У������Ե������� ʯ���ѡ�
������̼���ױ������ȼ��顢�ȷ¡����ᡢ������������ͪ���Ҵ����״���ˮ������ȡ�
��4���������ijߴ��Լ�������������Ҫ�Ӵ�������Ʒ�����ͷ������׳̶ȶ�����һ����
˵��������������������֮��ԼΪ8:1��������������ԼΪ��������Ʒ������30�����ҡ���
����װ�������Ժ�����Ӧ����Լ�ķ�֮һ�������������ܼ�����Ȼ�������Ʒ�������
�ѣ�����ѡ�ø���һЩ�IJ�������������������Ҳ���ʵ���һЩ��
��5���ܼ������ٶ����������Ч����������Ӱ�졣����ܼ����ٽ���������Ʒ�ڲ�����
�б�����ʱ��ͳ�����ô������ڹ̶����������֮����ܵõ���ֵ�������������ã���
��ʹ���������ǽṹ���������Ƶ���ֵ��Է��롣���ǣ��������������б�����ʱ��
̫������������ڸ�������ܼ��е���ɢ�ٶȴ������������ٶȣ��Ӷ�����ɫ�״��������
��ص�Ӱ�����Ч������ˣ�����ʱϴ���ٶ�Ҫ���С�
��6��װ��ʱҪ����ϵ��û����ӣ��Գ������ݣ������ѷ죬�����Ӱ�����Ч����
��7��װ����Ϻ��������������ܼ�ʱ��Ӧ�����ڻ������룬���⽫��������������Ʒ
�彦���𣬸����������������ɰ��Ҳ����������á�
��8������������������ڷ����ᴿ�⣬���������л�������ļ�����Ҳ��������Ѱ����
�����������������л��ϳ��У������������ٷ�Ӧ���̡������ԭ���ǣ����ñ�����ϵ���
������չ�����������е�ëϸ���ã�ʹ��Ʒ�������չ�����������������ڸ������������
���������ij̶Ȳ�ͬ���Լ���չ�������ܽ�ȵIJ��죬ʹ�������������еõ����롣һ�ֻ�
������һ�����������£��������߶���չ���������߶�֮����һ����ֵ����Ϊ�û�����ı�
��ֵ����ΪRfֵ�����������ȽϺͼ���ͬ���������Ҫ���ݡ�Ӧ��ָ������ʵ�ʹ����У�
Rfֵ�������Խϲ��ˣ��ڼ��������У�������֪���δ֪����ͬһ�鱡����ϵ�������
��ͬչ������ͬʱչ����ͨ���Ƚ����ǵ�Rfֵ�����������жϡ�
��9��������������õ��������й轺��������������ճ�ϼ��Ĺ轺�ƹ轺H������ճ��
������ʯ���Ϊ�轺G������ӫ�����ʵĹ轺��Ϊ�轺HF254�����ڲ���Ϊ 254 nm�������
�¹۲�ӫ�⣬�������ڹ�����ӫ�ⱡ���ϵ��л�������ȴ�ʰ�ɫ�ߵ㣬�����Ϳ��Թ۲쵽��
Щ��ɫ��֣��Ⱥ���ʯ���ֺ�ӫ�����ʵĹ轺��Ϊ�轺GF254��������Ҳ���Ƶط�Ϊ������G��
������HF254����������GF254��������ʯ���⣬�ȼ���ά����Ҳ�dz��õ�ճ�ϼ���������
�����ļ��Խ�ǿ�����ڼ������ʾ��н�ǿ���������ã�������ʺ��ڷ��뼫�Խ����Ļ�����
���������ѡ�±�����ȣ������轺�ļ�����Խ�С�����ʺ��ڷ��뼫�Խϴ�Ļ���������ᡢ
�������ȣ���
��10���ư�ʱ��һ��Ҫ�����������뵽�ܼ��У��ӱ߽��衣����ߵ����������ܼ��ӵ��������У����ײ�����顣
��11������ʱ������ëϸ�ܹܿ�Ҫƽ������������Ҫ������ݡ�������ʹ�ߵ��������β����ɢ������Ӱ�����Ч����
��12��չ�����ļ��Բ���Ի����ķ���������Ӱ�졣�������������ּ��Խ�ǿ���������������������и���ֵİߵ�ȫ�����ܼ���������ǰ�أ���ô���ܼ��ļ���̫ǿ���෴�����������и���ֵİߵ���ȫ�����ܼ���չ�����ƶ�������ܼ��ļ���̫����ѡ��չ����ʱ�����Բο��ڣ�3���Ӧ��ָ������ʱ�õ�һ�ܼ�����ʹ�������룬�����Ҫ���û���ܼ���չ���������ֻ��չ�����ļ��Գ����ڼ��ִ��ܼ��ļ���֮�䡣���Ѱ�Һ��ʵ�չ�����������·�������������һ�鱡չ���ϵ��ϴ�������Ʒ�ļ����ߵ㣬�ߵ������ 1cm���ϵļ�ࡣ�õιܽ���ͬ�ܼ��ֱ���ڲ�ͬ�İߵ��ϣ���Щ�ߵ㽫���ܼ����ܱ���չ�γɴ�С��һ��ͬ��Բ����ͨ���۲���ЩԲ���IJ�μ�࣬���ɴ����ж��ܼ��������ԡ�
��13������ɫ���ǹ۲���ɫ���ʰߵ��һ����Ч��������Ϊ��������������±��������Ĵ�����л����γ���ɫ�������������ڵ����������������ڿ����з���һ��ʱ�����ɫ�ߵ�ͻ���ʧ����ˣ���չ�徭����ɫ��Ӧ������Ǧ�ʽ���ɫ�ߵ�Ȧ�������������ϲ���ӫ�����ʣ����ֱ����������¹۲죬��������������������ʺ�ɫ�ߵ㡣
��14������ɫ������������Ϊ�����ࣨ����������һ��ɫ�������ݹ̶���״̬���ַ�Ϊ��-��ɫ������-Һɫ����ʵ�鷽���н��ܵ�����-Һɫ������-Һɫ�����Զ���Թ������������壨Ҳ�Ƶ��壩���������Ϳ��һ��ܱ��ĸ߷е�Һ���л���������Ϊ�̶��ࣨ�ֳƹ̶�Һ���������������ɫ�����С�����������������ɫ��������������ֽ��������̶�Һ֮�䷴�����з��䡣��Щ�ڹ̶�Һ���ܽ��С����ֺܿ�ͻᱻ�������������ڹ̶�Һ���ܽ�ȴ������ƶ��û������������ֱ����뿪������-��ɫ������-Һɫ��ԭ�����ơ�����������-��ɫ��������һЩ�������������轺��������������ֱ�����̶��ࡣ
��15������ɫ�����ͺźܶ࣬�����ǵ���ɻ�����ͬ����Ҫ����������Ӧϵͳ������ϵͳ��ɫ���������ϵͳ�Լ���¼ϵͳ�ȡ����������Ҫ�������û��Ͷ�����һ����˵����ɫ���ǿ����ȶ�������ע�������������������Ʒ����ɫ���������һ��������֣��������Ⱥ�������������������ЩŨ�Ȳ�ͬ�ĸ������Ӧ��ת��Ϊ���źţ����������ʽ��¼�ڼ�¼���ϡ�ͨ�������ӽ�����ʼ���������ij��ֵ�Ũ�����ֵ�����ʱ�䣬�Ʊ���ʱ�䡣һ����˵���л�����������ͬ�ķ��������£��䱣��ʱ���Dz���ġ���ˣ����Խ�������ɫ�������Է��������⣬����ֵĺ�����������������ȣ�������������С���ɽ��ж���������
��16��������ɫ�ײ���������Ҫ�õ��������м�����ע�ⰲȫ��
2.12�۹��ʵIJⶨ
�۹��ʣ�Refractive Index����Һ���л����������������֮һ��ͨ���ⶨ�۹��ʿ����ж�
�л�������Ĵ��ȣ�Ҳ������������δ֪�
2.12.1ʵ��ԭ��
�ڲ�ͬ�����У���Ĵ����ٶ��Dz���ͬ�ģ������һ�ֽ������뵽��һ�ֽ���ʱ���䴫����
��ᷢ���ı䣬����ǹ���������������䶨�ɣ������Խ���A�������B���������
��������Ǧµ�����֮�Ⱥ����ֽ��ʵ��۹��ʳɷ��ȡ�
sin��/ sin��= nB/ nA
���趨����AΪ������ʣ�����BΪ���ܽ��ʣ���nA<nB�����仰˵������Ǧ±�С������
�Ǧ�����ͼ2-26��
ͼ2-26 �������
�������Ǧ�=90������sin����1���������Ϊ���ֵ����Ϊ�ٽ�ǣ��Ԧ�0ʾ�����۹��ʵIJ�
�������ڿ����н��еģ����Կɽ��Ƶ����������״̬֮�У���nA=1�����У�
n=1/sin��0
��ˣ�ͨ���ⶨ�ٽ�Ǧ�0�����ɵõ����ʵ��۹���n��ͨ�����۹������ð�����Abbe���۹�
�����ⶨ���乤��ԭ�����ǻ��ڹ����������
���������IJ������ⶨ�¶ȵ����ض����ʵ��۹���������Ӱ�죬�����ⶨֵͨ��Ҫ��ע
�������������磬�� 20�������£����ƹ�D�߲�����589.3 nm���Ĺ��������������õ���
�Ȼ�̼���۹���Ϊ1.4600����ΪnD201.4600�������������ݿɶ���С��������λ����ȷ��
�ߣ��ظ��Ժã�������۹�����ΪҺ̬�л���Ĵ��ȱ������ȷе㻹Ҫ�ɿ������⣬�¶�
���۹��ʵ�Ӱ��ʷ��ȹ�ϵ��ͨ���¶�ÿ����l�棬�۹��ʽ��½�3.5��10-4��5.5��10-4��
Ϊ�˷����������ʵ�ʹ����г��� 4��10-4���Ƶ���Ϊ�¶ȱ仯���������磬���嶡����
�� 25��ʱ��ʵ��ֵΪ1.3670����У��ֵӦΪ��
nD20=1.3670+5��4��10-4=1.3690
2.12.2ʵ�鷽��
���۹��ǵ��⾵����ͼ2-27�������þ�ͷֽմ��ͪ�����⾵�ľ��棬Ȼ��� 1��2�δ�����Ʒ���⾵���ϣ������⾵����ת���⾵���ù����������⾵��ʹ������Ͳ�ӳ���������ת���⾵������ť��ֱ����Ŀ���пɹ۲쵽�����밵��ͼ���������ֲ�ɫ�����ɵ�����ɫɢ�⾵���⾵����ť����ʹ�����������������ţ��ٽ������ֽ��ߵ���������Ŀ���е�ʮ�ֽ��������غϣ���ͼ2-28������¼�������¶ȣ��ظ�2�Σ�ȡ��ƽ��ֵ���ⶨ��ϣ����⾵���ñ�ͪ�������档
ͼ2-27 �����۹���
2.12.3ע������
��1�����ڰ����۹�����������ɫɢ�⾵����ʹ��ɫ��ת��Ϊ��ɫ�⡣��ˣ���ֱ�������չ�ⶨ�۹��ʣ��������������ƹ�ʱ����õ�����һ����
��2��Ҫע�Ᵽ���۹��ǵ��⾵�����ɲⶨǿ���ǿ��Ⱦ߸�ʴ��Һ�塣
��3���ⶨ֮ǰ��һ��Ҫ�þ�ͷֽպ�����ӷ����ܼ����⾵������������������Һ�Ĵ��ڶ�Ӱ��ⶨ�����
��4������ⶨ�ӷ���Һ�壬�μ���Ʒʱ�����⾵�����С���롣
��5���ڲⶨ�۹���ʱ���������ͼ2-28��ʾ������ͼ2-28 ��4���Ƕ�ȡ����ʱ��ͼ����������ͼ2-28��1��������ɫɢ�������������⾵����ťֱ����ɫ�����ʧ��ͼ2-28��2��ͼ����Ȼ���ٵ����⾵������ťֱ����ͼ2-28��4��ͼ����������ͼ2-28��3��������������Ʒ���������£�����������Ʒ�����²ⶨ��
��6�����������Ͳ���ӳ�������Ӧ���С���⾵�Ƿ�����
ͼ2-28 �ⶨ�۹���ʱ�������
2.13����ȵIJⶨ
��ӳ���ǻ�Ϊ����������칹�塣���ǵ��۵㡢�е㡢����ܶȡ��۹����Լ������������ʶ���ͬ����������������Լ�����ʱ�����ǵĻ�ѧ����Ҳһ����Ψһ�ܹ���ӳ���ӽṹ��������������ǵ������Բ�ͬ����ƫ���ͨ�����й�ѧ���Ե�����ʱ��������ᷢ����ת������ת�ĽǶȼ�Ϊ����ȣ�Optical Rotation����
2.13.1ʵ��ԭ��
���������ʵ�����Ⱥ����ⷽ����������������ⶨ����������Ҫ��һ���ƹ�Դ��������ƶ��⾵��һ��ʢ�в�����Ʒ��ʢҺ����ɣ���ͼ2-29������ͨ���Ⱦ���һ���̶��������⾵����ƫ�������ƫ��⣬Ȼ��ͨ��ʢҺ�ܡ�����һ����ת�����⾵����ƫ����������ƫ�����������ת�Ƕȡ���ʹƫ�����ƽ��������ת�������������ʹƫ�����ƽ��������ת�����������
ͼ2-29 ������ʾ��ͼ
��������ʵ����������Ũ�ȡ������¶ȡ��Ⲩ����������������ء����ǣ���һ�������£�ÿһ�ֹ�������ʵ������Ϊһ�������ñ�����ȣۦ��ݱ�ʾ��
[��]��t = �� /(c��l)
���У���Ϊ�����Dz���ֵ��cΪ��Ʒ��ҺŨ�ȣ��� lmL��Һ������Ʒ������ʾ��lΪʢҺ�ܳ��ȣ���λΪdm����Ϊ��Դ������ͨ�������ƹ�Դ���� D��ʾ��tΪ�����¶ȡ����������ƷΪҺ�壬��ֱ�Ӳⶨ�����������Һ������������ʱ��ֻҪ��������ܶ�ֵ��d��������ʽ�е�Ũ��ֵ��C�����ɣ�
[��]��t = �� /(d��l)
���˱�������⣬�����ù�ѧ���ȡ������������ӳ��İٷֺ����Լ���ӳ�����ֵ��Enantiomer Excess,��дΪe.e����������ӳ��������ʵĴ��ȡ�
����SΪ�����칹�������е���Ҫ�칹�庬�������ӳ�����e.eֵ����ʽ���㣺
e.e.% = [(s-R)/(s+R)]��100%
���裨-����ӳ���ѧ����ΪX������
��-����ӳ��ٷֺ����� = [x+(100-x)/2]��100%
��+����ӳ��ٷֺ��� = (100-x)/2��100%
��ѧ���ȣ�P������Ϊ��
P =��[��] Dt��Ʒ/[��] Dt������100%
���磬��֪��Ʒ��S��-��-��-2-������������ܶ�d423 = 0.8����20cm����ʢҺ���У�������Ϊ -8.1�����������[��] = -5.8�������������У�
������� [��] Dt��Ʒ = ��23 /(C��l) = -8.1��/(2��0.8) = -5.1
��ѧ����P=��[��] Dt��Ʒ/[��] Dt������100% = (-5.1)/(-5.8) ��100% = 88%
(-)��ӳ��ٷֺ��� = [88+��100-88��/2] ��100% = 94%
(+)��ӳ��ٷֺ��� =��100-88��/2��100% = 6%
e.e.%=(S-R)/(S+R) ��100=88%
2.13.2ʵ�鷽��
�������ж������ͣ���������ʽ�Զ���ʾ������Ϊ����������������£�
��1��Ԥ�ȣ��������ǿ��أ�ʹ�ƵƼ��� 15 min������Դ�ȶ����ٰ��¡���Դ������
��2�����㣺��ʢҺ����װ���������ƴ�����Ʒ��Һ���ܼ�������ˮ����ʢҺ�ܷ����ڲ��Բ��е��㣬ʹ������ʾ������̶��̣�����Ϊ�㡣
��3��������Һ��ȷ��ȡ 0.l��O.5 g��Ʒ���� 25 mL����ƿ�������Һ��ͨ����ѡ��ˮ���Ҵ����ȷ����ܼ������ô�Һ����Ʒֱ�Ӳ��ԣ��ڲ���ǰȷ��������ܶȼ��ɡ�
��4�����ԣ�ѡ���ʵ����ȵ�ʢҺ�ܣ�����Ʒ��Һ��Һ����Ʒװ��ʢҺ���У�ע���ȥ���ݡ�Ȼ����ʢҺ�����������У����ϸǡ������ⶨ��������������ʾ������̶��̣������ȶ���������ٸ��⡢�������Σ�ȡ��ƽ��ֵ�����ݹ�ʽ���������ȡ���ӳ�����ֵ�ȡ�
2.13.3ע������
��1�������Ʒ�ı������ֵ��С�������ƴ�����Ʒ��Һʱ���˽�Ũ����ø�һЩ����ѡ�ó�һ��IJ���ʢҺ�ܣ��Ա�۲졣
��2���¶ȱ仯������Ⱦ���һ����Ӱ�졣�����ƹ⣨��=589.3 nm���²��ԣ��¶�ÿ����1�棬������������ʵ�����Ȼή��0.3�����ҡ�
��3������ʱ��ʢҺ�����÷ŵ�λ��Ӧ�̶����䣬�����������仯�������IJ�����
2.14�������
���ڷ��������˺����ߵ����������·��������ܼ���ԾǨ���Ӷ�������Ӧ�ļ�¼�źš���������ף�Infrared Spectroscopy�����IR����ͨ������������ж������л�������Ĺ����ţ������϶��ձ�������������Լ����л�������Ľṹ��
2.14.1ʵ��ԭ��
��������ǻ��ڷ�����ԭ�ӵ��������л����Ӳ��Ǹ��Խṹ�������й��ۼ�����һ������һ��Ƶ�ʵĺ��������»ᷢ��������ʽ�������������Ԧͱ�ʾ�����������Ԧı�ʾ���ȣ����������ַ�Ϊ�Գ��������Ԧ�s��ʾ���Ͳ��Գ��������Ԧ�as��ʾ������ͬ���͵Ļ��ϼ����������ǵ����ܼ���ͬ�������յĺ������ߵ�Ƶ��Ҳ��ͬ�����ͨ��������������Ƶ��ͼ�����������ͼ���Ϳ��Լ�����ֻ�ѧ����
����������ɺ�������Dz�ġ���������ǹ���ԭ����ͼ2-30��ʾ��
�������Դ���ɹ�̼����������̼���ڵ��������·��Ȳ������2��15�̷�Χ�������������⡣�����ⱻ���侵����ɿɱ䲨���ĺ���⣬����Ϊ������һ���Ǵ����ο��صIJαȹ⣻��һ����ͨ����Ʒ�ص����չ⡣
�����Ʒ��Ƶ�������仯�ĺ���ⲻʱ�ط���ǿ�Ȳ�һ�����գ���ô������Ʒ�ض����������������Ĺ�����ǿ�Ⱦͻ���Ӧ�ؼ�������������ǾͻὫ���չ�����αȹ������Ƚϣ���ͨ����¼�Ǽ�¼��ͼֽ���γɺ������ͼ��
ͼ2-30 ��������ǹ���ԭ��
���ڲ�����ʯӢ����������ȫ���ĺ���⣬��˲�����������Ʒ�ء�������Ʒ�صIJ���Ӧ���ǶԺ���������գ��Ա���������š����õIJ�����±�����Ȼ��ƺ��廯�صȡ�
2.14.2ʵ�鷽��
ͨ�����ⶨҺ����Ʒ�ĺ����������ҺĤ�����Ƚ�������Һ����Ʒ��һ������Ƭ�ϣ�������һ����Ƭ���ϣ���������ת������ʹ��ҺͿ�����ȡ�Ȼ��Ϳ��Һ����Ʒ����Ƭ�÷�����Ƭ֧���ϣ��������ں���������У���¼������ס�
������Ʒ�IJ���һ��ɲ���ʯ���ͣ�Nujol�����ƵĿ����ͣ��к�����±��ѹƬ����
ʯ�����к������� 3��5 mg���������Ʒ�� 2��3��ʯ�������в�����ĥ�ɺ�״��Ȼ��״��ͿĨ����Ƭ�ϲ�����һ����Ƭ���������档�ٽ�����Ƭ�÷�����Ƭ֧���ϣ��������ں���������У���¼������ס�
±��ѹƬ����ȡ 2��3 mg���������Ʒ���в�����ϸ���ټ��� 100��200 mg��ָ�������廯�أ������ĥ�ɼ�ϸ��ĩ��������װ�����ģ���С�������ģ�ߣ�ʹ�������ģ���зֲ����ȡ�Ȼ������������¼�ѹ��ʹ��ѹ��Ƭ״����ģ�ߣ�С�ĵ�ȡ����Ƭ���÷�����Ƭ֧���ϣ��������ں���������У���¼������ס�
2.14.3ע������
��l������ˮ�� 3710cm��1�� 1630cm��1 ��ǿ���շ壬����������������ʱ��������Ʒ����Ƭ�����ָ��ﴦ����
��2���� 5000��660 cm��1��Χ�ڼ�¼�������ʱ���˲����Ȼ�����Ƭ������ 830��400 cm��1��Χ�ڼ�¼�������ʱ���˲����廯����Ƭ��
��3��Ϊ�˷���������Ƭ��ͿĨ������Ʒʱ�����ں��������²�����������ϣ�Ӧ��ʱ�ö��ȼ�����ȷ²�ϴ�����������������б��á�
��4��ʯ����Ϊ̼�⻯����� 3030��2830cm��1�� C��H�������� 1460��1375 cm��1��C��H���������ڽ����������ʱӦע���Ƚ���Щ�廮ȥ�������ͼ����ȷ�����������š�
��5�������ؽ����������Ҫ�����ڻ��ۡ�ͨ�����ڷ���δ֪��ͼ��ʱ������Ҫ����Щ���ױ��ϵĻ����Ƿ���ڣ����ʻ����ǻ��������������˫���ȣ��Ӷ����Գ����жϷ��ӽṹ�Ļ����������������� 3000 cm��1����C��H�������շ��ؼ��ڷ�������Ϊ�������е��л��������ڸ����������ա����ڲ�ͬ����������е�ͬһ�����ں�������������ֵ�ϸ����Ҳ�������⡣δ֪�����ᆳ�������ṹ�����Ϳ��Բ��ı�ͼ���бȽϡ���Ϊ��ͬ�����������ͬ��ͼ�ף���ͺ���ͬ���˾��в�ͬ��ָ��һ������δ֪���ͼ�ͱ�ͼ����ȫһ��ʱ���Ϳ���ȷ��δ֪��ͱ�ͼ����ʾ������Ϊͬһ�����ͨ���ȽϽṹ����ĺ������ͼ��Ҳ���Ի��һЩ�вο���ֵ����Ϣ�����⣬�� 2000��1600 cm��1�� 1000��600 cm��1������ֵ��������������ȡ�������칹��ṹ��
2.15�˴Ź�����
�˴Ź����ף�Nuclear Magnetic Resonance��� NMR�����л���������ӽṹ�о�����һ����Ҫ���������ߡ��˴Ź���Դ���ܲ����ų��ĺ���������Լ�а���Ԫ�ص�ͬλ�ض������������ʡ����������л���ѧ�У���������Ҫ�ġ���ĺ˴Ź������Է�ӳ���л����ӽṹ�д��ڲ�ͬλ�õ���ԭ�ӡ������Ŀ�Լ��֮������ڹ�ϵ����Ϣ���ɴ˿��Ʋ������ӽṹ��
2.15.1ʵ��ԭ��
��ˣ�Ҳ�����ӣ���ͬС���壬�������÷��ڴų��У����ǽ����ų�����ȡ���˴Ź����������Բⶨ�ı�����ȡ������Ҫ����Ƶ��Ϊ�������˴Ź�������ԭ��ͼ��ͼ2-31������ʱ������Ʒ�ܲ�������
ͼ2-31 �˴Ź�������ԭ��
�����֮�䣬��Ʒ�ܵ������ϲ����Ž�����Ȧ���ڵ�����������ɨ����Ȧ������������Ȧ�ഹֱ�ķ����ϣ���������Ȧ����ͨ��������Ƶ����ͨ������Ȧ����Ʒ�������䡣
�����Ʒ����Ƶ������������Ƶ�ܲ������գ���Ϊ��Ƶ���������⣬���γɵ��źż�¼��ͼֽ�ϣ����ú˴Ź�����ͼ����ͼ�ϵ���λ�������ļ����飨TMS�����ź������㣬����Ծ����Ϊ��ѧλ�ƣ����Ԧ�����ȡ����ӵĻ�ѧλ��ȡ��������Χ�ĵ��ӻ���������ԭ�Ӻ���ת�ĵ�������ų������²����ĸ�Ӧ���ܶԿ���Ӵų�ʱ�����ӻ����������ö�ʹ��ʵ�ܴų����ͣ�������¹�������߳��ƶ�����ֵ��ý�С�����������ĸ�Ӧ�ų�����ǿ��Ӵų������ӻ���ȥ�������ã���ʹ��ʵ�ܴų���ǿ���Ӷ����¹�������ͳ��ƶ�����ֵ��ýϴ�ͨ���Ƚ�������ֱ�ӻ���������ԭ�ӵĵ縺�ԣ���һ���̶��ϾͿ��Դ���Ԥ�����������������õĴ�С������ RCH2����0.9����RNH2����1��5�������ڵ�ԭ�ӵĵ縺�Ա�̼ԭ��ǿ����ԭ�ӵ��������Ҫ��¶һЩ����������С������乲������ͳ�λ�ơ�����TMS�����еĹ�ԭ�ӵ縺�Ա�̼ԭ�ӵĻ�С�������TMS������Χ�ĵ����ܶȾߣ��������ô��乲����ͳ����ڸ��߳�����ʵ�ϣ���һ���л��������У���TMS����ЧӦ��ǿ�����Ӽ���û�У�������Ϊʲôѡ��TMS�������ԭ�������ӵĻ�ѧλ�Ʀ�ֵ��0��10֮�䡣
�ڽ����˴Ź�����ʱ��Ӧע�����¼��㣺
��1��������������Ӧ�ĺ˴Ź������շ���������ȡ��Լ��嶡����Ϊ����λ�ڦ�3.2�ĺ˴Ź����ź�������ԭ��ֱ�������ļ��ϵ����ӷ壬��1.2���ĺ˴Ź����ź����嶡���ϵ����ӷ塣�������֮��Ϊ1:3��������������������������֮�ȡ�
��2��n���������ӻ�ʹ�ڽ����Ӻ˴Ź����ź��ѷֳ�n��l�ط塣���磬�ڦ�-�����ѵĺ˴Ź������У�λ�ڦ�4.0�����ط�������ԭ��ֱ���������Ǽ��ϵ����ӷ壬λ�ڦ�1.5�������ط��Ǽ��ϵ����ӷ塣�������Ķ��ط�����������̼�ϵ����Ӽ䷢������ż�Ϻ������ѷֶ������ģ��Ҷ��ط��ǿ�ȱ���ѭ����չ��ʽϵ�����ڦ�-�����ѷ��ӽṹ�����Ǽ����ڵļ������������ӡ���ˣ��Ǽ������źű��ѷ�Ϊ���ط壬ǿ�ȱ�Ϊ1:2:2:1��ͬ��������������ڵ��Ǽ������������ӣ�����������źű��ѷ�Ϊ���ط壬ǿ�ȱ�Ϊ1:2:1��ֵ��ע����ǣ��ż�϶������ѷֵķ�������ȵģ�����Ҳ��ż�ϳ�������J��ʾ����λΪ���ȣ�Hz����ͨ���Ƚ�ż�ϳ���������Ѹ���б�ͬ�˴Ź����ź�֮��Ĺ�ϵ������ָ����ֻ������̼�ϵIJ����ԣ������ӻ�����ͬ�ģ����Ӽ�Ż��������ż�Ϻ������ѷ֡����ļ����������ķ��ӣ������Ӷ��ǵ��Եģ�����ᷢ��ż�����ã��������˴Ź�������ֻ����һ�����塣���嶡���ѵĺ˴Ź�����Ҳ���������Ρ���Ȼ�������л�������ĺ˴Ź����ױ���������Ҫ���ӵö࣬�ܶ�����»�Ҫ�������������Ϣ���й���������ѧ�������ۺϷ����ſɻ����ȷ�ķ��ӽṹ��
2.15.2ʵ�鷽��
��1��ѡ���ʵ��ܼ�����Ʒ�ܽⲢ���Ƴ�20�����ҵ���ҺԼ lmL��
��2������ܼ��в����ļ����飬��������Ʒ��Һ�м���1��2���ļ��������ڱꡣ
��3�������ƺõ���Ʒ��Һע�뵽ֱ��Ϊ 5mm����Լ18cm�IJ��Թ��У���Һע����������5 cm���
��4��װ����ϣ������ڽ�ʦ��ָ���½��в��ԡ�
2.15.3ע������
��l�������Ʒ��Һ̬������ֱ�Ӳ��ԣ������ƷΪ��̬������ճ�Խϴ��Һ̬���������Ƴ���Һ���в��ԡ�
��2��������Һʱ��Ӧѡ�ò������ӡ�������Ʒ������Ӧ���ܼ�������CCl4��CS2��DCCl3����Ϊ���Dz������ͼ�������š������Ʒ�������ܼ��ж����ܽ⣬����ѡ��D2O�����������ѡ뮴��ܼ�DCCl3��D2O����Ʒ�еĻ�����������ⷢ�������������Щ���ӵ��źŻ���ʧ����һ������ʱҲ�����������Լ��ס�Ӧ��ע�⣬�ڲ�ͬ���ܼ��в��ԣ��˴Ź����Ħ�ֵ����Щ�仯��
��3�����ѡ�� D2O���ܼ��������ļ����鲻�������У���ɲ��� 4��4-����-4-���������ƣ�TSPA����Ϊ���(CH3)3SiCH2CH2CH2SO3Na
2.16��ˮ������������
��Щ������ķ�Ӧ���Ժܸߣ����л��ϳ�������ʮ����Ҫ��Ӧ�ã�Ȼ�����ǶԿ�����ˮ��Ҳ�dz����У�����Ʊ���������˽ϸߵ�Ҫ��ͨ����Ҫ����ˮ�����������Ͻ��в�����
2.16.1ʵ��ԭ��
��ˮ����������Ҳ��ʷ�����ߣ�Schlenk Line������һ��������ľ���������ϵͳ��ͨ������ϵͳ�����Խ���ˮ�����������嵼�뷴Ӧϵͳ���Ӷ�ʹ��Ӧ����ˮ����������˳�����С���ˮ������������Ҫ�ɳ���������������Na��K�Ͻ�ܡ����ܡ�˫�Źܡ���ռƵȲ�����ɣ���ͼ2-32��ʾ��
ͼ2-32 ��ˮ����������
�������壨�������������һ��ѹ�����ɹ��������밲ȫ�ܾ�������������ˮ���ٽ���������Գ�����Ȼ�����ڶ��������������ճ����������ɵ���ˮ���̶�ͨ��Na��K�Ͻ���Գ�ȥ������ˮ������������ܽ���˫�Źܣ������������ܣ���
�ڸ������У��������ˮ����ǿ���������ĸ�������� 5A����ɸ���ڳ���������ѡ�ó���Ч���ò��������ij���������������ɸ��������������ˮ����ϵͳ������Ķ������壬�Ϳ��Ե��뵽��Ӧϵͳ����������ϵͳ��
2.16.2ʵ�鷽��
��ʹ����ˮ����������֮ǰ������Ҫ�Ը������ͳ��������л��
��ѡ��5A����ɸ������������ڳ�Ϊ60cm���ھ�Ϊ3cm�IJ������У�װ��5A����ɸ���������϶˲�������Ϊ 400����¶ȼƣ��������� 500 W����˿�����������ϳ�Ϊ 60 cm���ھ�Ϊ 6 cm�IJ����ܡ������¶�����ͨ���ֱ�����ձü�����������ӡ��� 1.33 kPa��10mmHg����320��350��������¶Է���ɸ��� 10 h��Ȼ����ת��ͨ������������壬ֹͣ���ȣ���Ȼ��ȴ�����£�����������������ϵͳ��
��ѡ��������ɸ�����������ڳ�Ϊ60 cm���ھ�Ϊ3 cm�IJ������ڣ�װ��������ɸ�������϶˲�������Ϊ 400����¶ȼƣ��������� 300 W�ĵ���˿�����������ϳ�Ϊ 60 cm���ھ�Ϊ 6cm����ܡ��ʱ�������¶˲��ͨ��������β�������϶˲��ͨ�����⡣������90��110�棬� 10 h���ң�����������ɵ�����ˮ����ͨ�����¶˵ĵ��ܷų�����������ɸ��ں�ֹͣ���ȣ�����ͨ��������Ȼ��ȴ�����£����ϸ�������������ϵͳ��Na��K�Ͻ���϶˳�Ϊ50cm���ھ�Ϊ 2 cm���¶˳�Ϊ 15 cm���ھ�Ϊ 5 cm���϶˲������ͨ�����ֱ�����ձúͶ���������ӡ��ȳ���ղ��õ紵���ú���ƺ濾����Ȼ��ȴ�����£��ٳ��
�����壬�黻�����Ρ��ڳ�������������£����Ͽڼ���������ƣ�15 g���ͼأ�45 g��������������ʯ���ͼ��Ը��ǡ�Ȼ������¶ˣ�ʹ�ơ������ڣ���ȴ��Na��K�Ͻ𡣲����ѳ黻�����ڹܣ�����������������ϵͳ��
���������Ӵ������������Ϳ��Խ��г�ˮ����������
��Ҫ���ˮ����������ͨ���������ĵ��ܣ�����ˮ�����������ϵ�˫�Ź������Ա�黻�����ڸ�������֧�ڴ�Ҫ����Һ����Ա�ſա�ͬʱ���������ڶ�������Ϊ��ѹ��ʹ�����������ڡ��ر�֧�ڴ���Һ��ܣ���ת˫�Źܵ�˫б����ʹ��ϵ����չ�����������գ��õ紵���ú���ƺ濾������ϵͳ�����֣��Գ�ȥϵͳ�ڵĿ������ڱڸ��ŵij������濾��ϣ���������ȴ�������巧����ת˫�Ź���˫б��ͨ��ʹ������ϵͳ����������·��ͨ���������ظ�����3�Σ����黻����ϡ�
�ڶ��������½��и��ֲ�����װ�ü�ͼ2-33ϵ�С�
2.16.3ע������
��1���������Ҫ���� 2 mL��m3�ķ�Χ����ʷ���˲������Ͽ��Բ���Na��K�Ͻ�ܡ�
��2���� 5A����ɸ������������壨�����������������������ˮƽ������ѹС�� 0.13 Pa��
��3����������ɸ�������ף������ϴ�������һ�㾭������ɸ����������Ķ������壬�京�����ɽ��� 2 mL��m3���¡�
��4����ˮ���������������ý����˲��ú����Ƥ�ܣ��Է��黻��ʱ�п������롣
��5������ڷ�Ӧ������Ҫ����ҩƷ�������������Ҫ������Ӧƿʱ����Ӧ�ڽϴ�Ķ��������н��в�����
��6����Ӧϵͳ������裬Ӧʹ�ô������衣��ʹ�û�е��������Ӧ�Ӵ����������������
��7����Ҫ�����ѡ�����ૡ��ױ����ܼ����ϸ���ˮ�����������ɰ����²�����У�������װ��ͨ����ͨ������ˮ�������������������黻��������˿Ԥ���������ܼ��Լ�ͭ��Ͷ�����ͪ����l:4�����ȣ�ת�����С���ת˫б��ͨ������ʹ������ͨ���ֻ���������Һ�ɻ�ɫ��Ϊ����ɫ���ɹ���˫б��ͨ��ʹ�ܼ���������Һǻ�У����ܼ��е�ˮ�ֺ����������������Ʊ㽫������ͪ��ԭ�ɱ�Ƭ�Ŵ��ƣ��ʳ�����ɫ����ȡ�ܼ�ʱ����ע�������Ͽڳ������ת˫б��ͨ���²�ܷų���
��8����ˮ�����������У���������װ��ʯ���ͺ���ͨ����������һ������Է���ع۲���ϵ�ڶ��������������������һ����Ҳ��������ϵ�ڲ�ѹ�����¶����仯������ѹʱ��ʹ�ڲ����ⲿ��������ֹ�������롣ˮ����ȫ�ܵ�������Ҫ��Ϊ�˷�ֹ��Ӧϵͳ�ڲ�ѹ��̫������½�ƿ���忪�����ȿ��Ա���ϵͳһ����ѹ�����ֿ�����ϵͳѹ������ʱ���ö���������зſա�����ƿ���Ų����������д�����ʯ���͵����á�������װ�л�ķ���ɸ��
�����ն����������ٹ���ʱ����-�غϽ���д���������ʯ���ͣ�������뷴Ӧ����
��9���ڳ�����Ӧ�У����������ˮ��������Ҫ���Ǻܸߣ�ֻҪ����һ���������������������Ϳ����ˡ�
ͼ2-33 �ڶ��������½��и��ֲ�����װ��
2.17���ȡ����估���\��
2.17.1���ȷ���
������ʹ�л���Ӧ���١�ͨ������Ӧ�¶�ÿ���10�棬��Ӧ�ٶȾͻ�����һ�������õļ��ȷ�ʽ�п���ԡ��ˮԡ����ԡ��ɰԡ��
��1������ԡ
ֱ������ú���Ƹ���ʯ�����Բ����������ȼ�Ϊ����ԡ������������ʯ����Լ 1cm��ʹ�м��϶��ʯ�����µĻ���������ȿ��������ּ��ȷ�ʽ�����ң���ʮ�־��ȣ�������ʺ��ڵͷе���ȼҺ��Ļ���������Ҳ�������ڼ�ѹ�����������ú�����⣬������Ҳ�����ڿ���ԡ���ȣ���ͼ2-34����
��2��ˮԡ
����Ӧ��������ˮԡ���У�ʹˮԡҺ���Ը߳���Ӧ�����ڵ�Һ�棬ͨ��ú���ƻ��������ˮԡ�����ȣ�ʹˮԡ�¶ȴﵽ�����¶ȷ�Χ�������ԡ������ȣ�ˮԡ���Ⱦ��ȣ��¶����ƣ��ʺ��ڵͷе����ʻ������ȡ�
��������¶Ƚӽ�100�棬���÷�ˮԡ��ˮ����ԡ��Ҫע����ǣ�����ˮ����������ڲ��������У�Ӧ��ʱ��ˮ����ͼ2-35����
ͼ2-34 �������ȵĻ���װ��ͼ 2-35 ˮԡ���ȵĻ���װ��ͼ
��3����ԡ
�������¶���100��250�淶Χ��Ӧ������ԡ�����õ���ԡԡҺ��ʯ���͡����͡���ձ��ͻ�һЩֲ���ͣ��綹�͡������͡������͵ȡ�����ԡ����ʱ������ע���ȡ��ʩ����Ҫ��ˮ�������У��������ʱ�������ĭ������ɽ������磬�ڻ����������¶�����һ����ֽȦ���������µ�ˮ�Ρ���ʹ��ֲ����ʱ������ֲ�����ڸ����������ֽ⣬�������м���l���Ա����ӣ������������ȶ��ԡ����ͺ���ձ��ͼ����¶ȶ��ɴﵽ250�棬���ȶ��Ժã�ֻ�Ǽ۸�Ϲ�
��4��ɰԡ
�������¶���250��350�淶Χ��Ӧ����ɰԡ��ͨ����ϸɰװ�������У��ѷ�Ӧ��������ɰ�У���������ײ�����һ��ɰ�㣬�Է��ֲ����ȡ�����ɰԡ�¶ȷֲ������ȣ��ʲ���ԡ�µ��¶ȼ�ˮ����Ӧ������Ӧ������
2.17.2���䷽��
����һЩʵ��Ե��µ�Ҫ���ڲ�������Ҫʹ������������磬����һЩ���ȷ�Ӧ��������
��Ӧ�����У��¶Ȼ�����ߣ�Ϊ�˱��ⷴӦ���ھ��ң����Խ���Ӧ������û����ˮ�л��ˮ�У����ˮ�Է�Ӧ��Ӱ�죬�����Խ�����ֱ��Ͷ�뵽��Ӧ�����н�����ȴ�������Ҫ���͵��¶ȣ�����0�棩�����Բ��ñ�-�λ��������ȴ������ͬ���κͱ���һ���������Ƴ������¶ȷ�Χ��ͬ����ȴ��������1��
��1 ������ȴ����ɼ������ȴ�¶�
��ȴ�����
�����ȴ�¶ȣ��棩
�Ȼ��+�����1 ��4��
-15
�Ȼ���+�����1 ��3��
-21
��ˮ�Ȼ���+�����1 ��1��
-29
��ˮ�Ȼ���+�����1 ��4 ��1��
-55
�ɱ�+�Ҵ�
-72
�ɱ�+��ͪ
-78
�ɱ�+����
-100
��ˮ
Һ��
-196
Ӧ��ע�⣬��������¶ȵ���-38�棬����Ӧ������װ�л�Һ��ĵ����¶ȼƣ�������ʹ��ˮ���¶ȼƣ�ˮ�������̵�Ϊ-38.9�棩��
2.17.3���﷽��
����ָ���dz�ȥ���塢Һ��������е�ˮ�֡��л��������������Բ��ԡ����뷴Ӧ������ǰ��Ҫ���и��ﴦ�������ݳ�ˮԭ�������﷽���ɷ�Ϊ���������ͻ�ѧ������
��������������������������������ȣ�Ҳ�ɲ������ӽ�����֬�����ɸ��ˮ�����ӽ�����֬�ͷ���ɸ�����������ˮ�Թ��壬���Ⱥ��ֻ��ͷų�ˮ���ӣ��ʿɷ���ʹ�á�
��ѧ������ˮ��Ҫ�����ø������ˮ�ַ���������淴Ӧ����ˮ�����磬��ˮ�Ȼ��ơ���ˮ����þ������ˮ��Ӧ�����������ˮ�������һЩ�����������ơ����������ȿ���ˮ���������淴Ӧ�����µĻ����
��1��Һ���л�������ĸ���
һ��ɽ�Һ���л������������״���������һ�����ķ�ʽ���и��ﴦ��������л��������к�ˮ���ϴɷִν��и��ﴦ����ֱ�����¼���ĸ�������������Ե���ˮ����Ϊֹ�����磬�Ȼ����Ա��ֿ���״�����������ײ��ٽ��ȡ�ѡ����ʸ������ԭ���ǣ����뱻���ﻯ�������ѧ��Ӧ�����ܽ��ڸû������ˮ���ϴ����ٶȽϿ죬���Ҽ۸���������ø���������ܼ�Ӧ�÷�Χ����2��Һ���л�����������ø�����⣬���ɲ��ù�������ķ�����ˮ��
��2�������л�������ĸ���
��������л�����������ķ������ǽ���̯���ڱ��������ֽ����Ȼ���ɣ�������ֻ�ʺ��ڷ���ʪ�Ի����������������ȶ��Ժã����۵�ϸߣ��Ϳɽ������ں����л������½��к�ɴ�����������Щ������������ʱ�ֽ�Ļ��������÷��ڸ������н��и��
��2 ���ø������Ӧ�÷�Χ
�������
�����
��
±��
��
��
ȩ
ͪ
��
����������
�л��ᡢ��
��
�Ȼ��ơ������ơ�����ɸ
�Ȼ��ơ�����þ��������
̼��ء�����þ�������ơ�������
����þ��������
����þ��������
̼��ء��Ȼ��ƣ���ͪ�����ã�
����þ�������ơ��Ȼ��ơ�̼���
�Ȼ��ơ�����þ��������
����þ��������
�������ơ��������ء�̼���
2.17.4ע������
��l�� CaCl2��ˮ�����ٶȿ죬���������������ڴ��������ӡ������ᡢ�����ȡ�
��2��Na2SO4��ˮ������������Ч���ͣ�����Ϊ�����������
��3��MgSO4��ˮ������Na2SO4�ȣ����ÿ죬Ч���ߡ�
��4��K2CO3���ڼ��Ի����������������ᡢ�ӵ����Ի����
��5��Na�������ѡ��尷�����к���ˮ�ĸ�����������ȴ��������������Խ��������еĻ����
ʵ��һ �����������Ͳ�����������֪ʶ
һ��ʵ��Ŀ��
1����ʶ��ĥ�ڲ�������������;
2����ϰ�����ܵļӹ�����ȷ�����л���ѧʵ����������������˽����������л���ѧʵ��ij���װ�ã��˽�ӷ�Ӧ��ϵ�з��롢�ᴿ�ͼ���Ŀ�껯����ķ�����
����ʵ��������ҩƷ
ϵ�б�ĥ�ڲ�������������ﱵ�������ɰ�֣����ƾ���ơ������ܡ�ʯ�������ƾ�
����ʵ������
1. �����ܵĽض�
a��ﱺۣ��Ѳ�����ƽ�������ӱ�Ե�ϣ���Ĵָ��סҪ�ضϵĵط���������ﱵ���ߣ�����ɰ�֣�����ﱳ�һ�����ۣ�Լռ����1/6��ﱺ�ʱֻ����һ��������ǰ������ȥ������������ﱡ�
b���۶ϣ����ֱַ���ס���۵����ߣ��������⣬������Ĵָ�ֱ�ס���ۺ�������࣬������������һѹ�������۳����Ρ�
2. ����������
˫�ֲֳ����ܣ������������Ҫ�����ĵط����ڻ���������Ԥ�ȣ�Ȼ������β���ϼ��ȡ��ڻ�����ʹ�����ܻ��������ȡ�������ͬһ������ת�������������ȣ���ƣ����ӻ�����ȡ�����ڻ����ϼ��Ⱦ�����Ҫ���������������������Ҫ�ĽǶȣ������Ҫ�Ƕ�֮���ڹܿ����ᴵ�������ֹ��ھ����ȣ�Ȼ�����ʯ��������Ȼ��ȴ��
3. �ιܡ�ëϸ�ܺ��۵�ܵ�����
�ιܡ�ëϸ�ܵ����ƣ�������������ϣ����ֵĴ�Ĵָ��ʳָ����ָ���Ų��������ˣ�������ԣ��ڻ����ϼ��ȣ���ͣ������ͬ����ת�������������ճɺ��ɫʱ���ӻ�����ȡ��������ƽ�ȵ���ˮƽ�������෴�����ƶ�����ʼʱ��Щ��Ȼ��ӿ죬��ȴ�����۶ϴ���ɰ�ֻ��ۣ��۶Ͼͳ������ιܣ���Ѵ�ڶ��û����ճɾ��ߣ��ͽϳ�һ���ھ�ԼΪ1mm��ëϸ�ܡ�
�۵�ܵ����ƣ����ھ�1mm��ëϸ�ܽس�7-8cm������С���Ͼ��ȷ��һ�ˣ�ע����������������С�ȡ����汸�á�
4. ���ñ�������������ʶ�Լ�ʹ��
������������ú��л���ѧʵ���ҳ�����ͨ�����������ر��dz��ñ�ĥ�ڲ������������ҽ�����Щ������ʹ�÷�������ϴ�����﷽����
��ѧ�������˽��л���ѧʵ���г��ó��ײ����豸�İ�װ��ʹ�úͲ�ж�������������Ӧװ�á���ѹ����װ�õȡ�
������Ӧװ�� ��ѹ����װ��
�ġ���������
1. �ضϲ�����ʱ��Ҫע����Щ���⣿������������ϸ�����ܣ��ڻ����ϼ��Ȳ�����ʱ�������ܷ�ֹ�����ܱ����
2. ��������ϸ������ʱ���������ܵ��¶���ʲô��ͬ��ΪʲôҪ��ͬ�أ����ƺõ������������Ҫ�������������Ӵ��ᷢ��ʲô�����ĺ����Ӧ���������ܱ��⣿
ʵ��� �۵�IJⶨ
һ��ʵ��Ŀ��
�˽��۵�ⶨ�����壻����ëϸ�ܷ��ⶨ�۵�IJ����������˽��۵��ǵ�֪ʶ��
����ʵ��ԭ��
�۵��ǹ����л�������̡�Һ��̬�ڴ���ѹ���´��ƽ����¶ȣ������Ĺ����л�������һ�㶼�й̶����۵㣬�̡�Һ��̬֮��ı仯�Ƿdz�����ģ��۳̣��Գ�����ȫ�ۣ�һ�㲻����0.5-1�档���ȴ������л�������ʱ�����¶������ʱ��ı仯����ͼ��
ͼ1 �������廯��������ʱ����¶ȵı仯
���������廯�����¶Ȳ����۵�ʱ�Թ�����ڣ�����ʹ�¶��������ﵽ�۵㡣��ʼ������Һ����֣������Һ��ƽ�⡣�������ȣ��¶Ȳ��ٱ仯����ʱ�������ṩ������ʹ�����ת��ΪҺ�࣬�������Ϊƽ�⣬���Ĺ����ۻ������������¶���������������ڽӽ��۵�ʱ�������ٶ�һ��Ҫ����ÿ�����¶����߲��ܳ���2�棬ֻ������������ʹ�����ۻ����̾����ܽӽ�������ƽ����������õ��۵�ҲԽ��ȷ��
��������ʱ���ٶ����߲��γɹ����壩���������ڶ����ɿ�֪����һ����ѹ�����¶������£����ܼ������ӷǻӷ����ʺ��ܼ�������ѹ���ͣ�ͼ2��M��L�䣩����Һ���ཻ��M�伴�����������ʻ�����ﵽ�۵�ʱ�Ĺ�Һ��ƽ���㣬TM��Ϊ������ʱ���۵㣬��Ȼ����ʱ���۵�ϴ�����������۵�ͣ��۳̳���ͼ3����
ͼ2 ��������ѹ���¶ȱ仯���� ͼ3 ���������廯��������ʱ����¶ȵı仯
����ҩƷ������
ҩƷ�������ͣ����ͣ��� �����ᣬ ���������� ���� δ֪��
�������¶ȼƣ� B��(Thiele��)
�ġ�ʵ�����
1����Ʒ��װ��
��������Ʒ���ڸ���ྻ�ı��������ϸ�Ҽ���һС�ѡ��۵�ܿ���һ�˴�ֱ���˶Ѽ�����Ʒ�У�ʹһЩ��Ʒ������ڣ�Ȼ���۵�ܿ������Ϸ��볤Լ50��60cm��ֱ����IJ������У����µ������ʹ֮�Ӹߴ����ڱ������ϣ���˷������Σ��ɰ���Ʒ��ʵ����Ʒ�߶�1-2mm�������۵�������Ʒ��ĩ������Ⱦ����Һ�塣
2�����۵�
��ͼ2-16��ʾ��װ��װ�ã�������ͣ� С�ĵؽ�װ����Ʒ���۵��ճ����պȡ�������͵�ˮ������ϣ����ȡһС����ƤȦ�����¶ȼƺ��۵�ܵ��ϲ�����ճ�����۵�ܵ��¶ȼ�С�ĵز���(Thiele���е���ԡ���С����ͼʾ��λ���ȡ���ʼ�����ٶȿ�Щ�����������¶ȵ��ڸû������۵�10��15��ʱ����������ʹÿ��������Լ1��2�棬���ӽ��۵㣬�����ٶ���Ӧ������ÿ����Լ0.2��0.3�档 ������Ʒ���ۺ�ȫ�۵��¶ȶ�������Ϊ�û�������۳̡�Ҫע���ڼ��ȹ����������Ƿ���ή������ɫ�����ݡ�������̼��������Ӧ��ʵ��¼��
�ظ��ⶨ2��3�Ρ�ÿһ�βⶨ�������µ��۵����װ��Ʒ��
����ⶨδ֪����۵㣬Ӧ�ȶ���Ʒ�ֲ�һ�Σ����ȿ����Կ죬֪�����µ��۾ࡣ��ԡ�������۵�����30�����ң�����ȡһ��װ���������۵����ȷ�IJⶨ��
3���¶ȼ�У��
���۵�ʱ���¶ȼ��ϵ��۵��������ʵ�۵�֮�䳣��һ����ƫ��������������ԭ�����ȣ��¶ȼƵ������������ëϸ�������ȣ��̶Ȳ�ȷ����Σ��¶ȼ���ȫ��ʽ�Ͱ��ʽ���֣�ȫ��ʽ�¶ȼƵĿ̶������¶ȼƹ���ȫ���������ȵ�����¿̳����ģ������۵�ʱ���в��ֹ������ȣ����¶���Ĺ����¶Ƚ�ȫ�������ߵ͡�Ϊ��У���¶ȼƣ���ѡ�ô��л���������۵���Ϊ����ѡ��һ���¶ȼ�У����
ѡ��������֪�۵�Ĵ�������Ϊ���ƣ��ⶨ���ǵ��۵㣬�Թ۲쵽���۵��������꣬����۵�����֪�۵��ֵ�������꣬�������ߣ����ɴ������϶�����һ�¶ȵ�У��ֵ��
���ñ���Ʒ����1��
��Ʒ����
�۵㣨�棩
��Ʒ����
�۵㣨�棩
ˮ ~ ��
����
132
�Զ��ȱ�
53
ˮ����
159
�����ᱽ��
70
2,4-������������
183
��
80
3,5-������������
205
���������
90
��
216
��������
114
������������
242
������
122
����
286
�塢ʵ��ע������
1���۵�ܱ���ྻ���纬�лҳ��ȣ��ܲ���4��10�����
2���۵�ܵ�δ��û����©�ܡ�
3����Ʒ����Ҫϸ����װҪʵ�����������϶�����״��ȣ�����۳̱��
4����Ʒ������������ʣ���ʹ�۵�ƫ�ͣ��۳̱��
5����Ʒ��̫�ٲ���۲죬�����۵�ƫ�ͣ�̫�������۳̱���۵�ƫ�ߡ�
6�������ٶ�Ӧ�������ȴ����г�ֵ�ʱ�䡣�����ٶȹ��죬�۵�ƫ�ߡ�
7���۵�ܱ�̫���ȴ���ʱ�䳤��������۵�ƫ�ߡ�
8��ʹ������������ԡҺҪ�ر�С�ģ��������л�������Ũ���ᣬ����ʹԡҺ��ɫ����а��۵�Ĺ۲졣����������������ɼ�����������ؾ��干�Ⱥ�ʹ֮��ɫ������Ũ��������ԡ�������ڲ��۵���220�����µ���Ʒ����Ҫ���۵���220�����ϵ���Ʒ����������ԡҺ��
����˼����
���۵�ʱ�������������������ʲô�����
��1���۵�ܱ�̫��
��2���۵�ܵײ�δ��ȫ��գ�����һ��ס�
��3���۵�ܲ��ྻ��
��4����Ʒδ��ȫ����������ʡ�
��5����Ʒ�еò�ϸ��װ�ò����ܡ�
��6������̫�졣
ʵ���� ���е�IJⶨ
һ��ʵ��Ŀ��
1����Ϥ��ѹ����ͳ������ⶨ�е��ԭ�����˽�����Ͳⶨ�е�����壻
2����������Ͳⶨ�е�IJ���Ҫ��ͷ�����
����ʵ��ԭ��
Һ��������ڷ����˶��дӱ����ݳ��������������������¶ȵ����߶���������Һ���ϲ��γ���������������Һ���ݳ����ٶ�������������ص�Һ���е��ٶ����ʱ��Һ���ϵ������ﵽ���ͣ���Ϊ��������������Һ����ʩ�ӵ�ѹ����Ϊ��������ѹ��ʵ��֤����Һ�������ѹֻ���¶��йأ���Һ����һ���¶��¾���һ��������ѹ��
��Һ�������ѹ���������ʩ��Һ�����ѹ����ͨ���Ǵ���ѹ�������ʱ�����д������ݴ�Һ���ڲ��ݳ�����Һ����ڣ���ʱ���¶ȳ�ΪҺ��ķе㡣
������Һ���л���������һ��ѹ���¾���һ���ķе㣨�г�0.5-1.5�棩��������һ�㣬���ǿ��Բⶨ��Һ���л���ķе㡣�ֳƳ�������
���Ǿ��й̶��е��Һ�岻һ�����Ǵ���Ļ������ΪijЩ�л������ﳣ����������γɶ�Ԫ����Ԫ���л�������Ҳ��һ���ķе㡣
�����ǽ�Һ���л�����ȵ�����״̬��ʹҺ�����������ֽ���������ΪҺ��Ĺ��̡�
ͨ������ɳ�ȥ���ӷ������ʣ��ɷ���е�����30 oC��Һ����������Բⶨ��Һ���л���ķе㼰���Լ���Һ���л���Ĵ��ȡ�
����ҩƷ������
ҩƷ�� �Ҵ�
������ ����ƿ������ͷ���¶ȼƣ�ֱ�������ܣ�β�ӹܣ� ��ƿ����Ͳ
�ġ�ʵ��װ��
Ҫ�������������ͽ������������ ������ͼ��ʾ��
1������ƿ������ƿ��ѡ���뱻��Һ�����Ķ����йأ�ͨ��װ��Һ������ӦΪ����ƿ�ݻ���1/3-2/3��Һ�����������ٶ����ˡ�
2������ͷ��������ͷе�Һ��ʱ��ѡ�ó�������ͷ��������߷е�Һ��ʱ��ѡ�ö̾�����ƿ��
3���¶ȼƣ��¶ȼ�Ӧ���ݱ�����Һ��ķе���ѡ�� ���ݾ�ȷ�ȵ�Ҫ���Һ��е�ߵ�ȷ���¶ȼƵ�ѡ�á�
3�������ܣ������ܿɷ�Ϊˮ�����ܺͿ������������࣬ˮ���������ڱ���Һ��е����140 oC���������������ڱ���Һ��е����140 oC��
4��β�ӹܼ�����ƿ��β�ӹܽ�����Һ�������ƿ�С���ѹ����ѡ����ƿΪ����ƿ����ѹ����ѡ��Բ����ƿΪ����ƿ��
������װ˳��Ϊ�����º��ϣ�������ҡ�ж��������˳���෴��
�塢ʵ�鲽��
1�����ϣ��������Ҵ�40mLС�ĵ�������ƿ�У���ҪʹҺ���֧��������������������ӣ��Ӻ�����ͷ�����ô��¶ȼƵ����ӣ�ע���¶ȼƵ�λ�á����װ���Ƿ������������ԡ�����������������
2�����ȣ��ȴ�����ˮ��ͷ��ע����ˮ���¶��ϣ�����ͨ����ˮ��Ȼ��ʼ���ȡ� ��Һ����ڣ���������ˮ����λʱ���¶ȼƶ�������������������Դ����ˮ������Һ�κ������¶ȴﵽƽ�⣬ʹ�����ٶ���ÿ��1��2��Ϊ�ˡ���ʱ�¶ȼƶ����������Һ�ķе㡣
3���ռ���Һ������������ƿ��һ������ǰ��֣���һ��������أ�����������֣������¸���ֵķг̣�������ֵĵ�һ�κ����һ��ʱ�¶ȼƵĶ�����
����������������¶ȼƶ�����ͻȻ�½�����ʱӦֹͣ����ʹ���ʺ��٣�Ҳ��Ҫ���ɣ���������ƿ���Ѽ��������������¹ʡ�
4���������װ�ã�������ϣ���Ӧ������Դ��Ȼ��ֹͣͨˮ�����������װ�ã��밲װ˳���෴����
����ʵ��ע������
1����ȴˮ�������ܱ�֤�����������Ϊ�ˣ�ͨ��ֻ�豣�ֻ���ˮ�����ɡ�
2�������л��ܼ���Ӧ��С�ڽ�����������ƿ��
3���ڿ�ʼ����ǰ��������ʯ���������ɶ�ף������������˵�dz���Ҫ����Ϊ�������Һ���ڼ���ʱ�����������������¶ȳ����е�Ҳ�����ڣ�����������ȣ�Һ��ͻ������������������ƿ�⡣��ʯ����������������һЩ�������ڼ���ʱ�Ϳ����γ�Һ��������������ģ��Ӷ���֤Һ�弰ʱ���ڶ����ⱬ�С��������ǰ���˼ӷ�ʯ������ʱӦֹͣ���ȣ���Һ�������е����ºɼ��롣����������;ֹͣ������������ʱӦ�����µķ�ʯ��
4������ʱ���ܼ���̫�죬�����������ƿ�ľ�����ɹ�������ʹˮ����������������������������¶ȼƶ��õķе�ƫ�ߣ���һ��������Ҳ���ܽ��е�̫�������������¶ȼƵ�ˮ������Ϊ���Һ�����������ʹ�¶ȼ��������õķе�ƫ�͡�
�ߡ�˼����
1��ʲô�зе㣿Һ��ķе�ʹ���ѹ��ʲô��ϵ����������ص�ij���ʵķе��Ƿ�Ϊ��������ķе��¶ȣ�
2��Ϊʲô����ʱ��ÿ������Һ���ٶ�Ϊÿ��1��2��Ϊ�ˣ�
ʵ���� Һ̬�л��������۹��ʵIJⶨ
һ��ʵ��Ŀ��
1��ѧϰ�л��������۹��ʵIJⶨ��
2��ѧϰAbbe�۹��ǵ�ʹ�÷�����
����ʵ��ԭ��
�������䶨�ɣ�n=
��������������������߲�����ͬ���䣬Ҳ���¶Ȳ�ͬ���䣬�¶�ÿ����1�棬�۹����½�
��3.5-5.5����10-4������nDt����ʾ��Һ�����ʾ����ض����۹��ʡ�
����ʵ��������ҩƷ
Abbe�۹��ǡ���������ˮԡ������ͪ���Ҵ�������ֽ
1��������Զ�� 2��������Զ��
3����ɫɢ���ֱ� 4������ˮ���
5���¶ȼ� 6�������⾵ 7������
8�������⾵������״̬�� 9����Һ��
10�����侵 11������ 12����ť
13��ת�� 14���̶�����
�����۹��ǵĽṹ
�ġ�ʵ�鲽��
1������Abbe�۹��ǵĹ��죨1.3000-1.7000��
2�����ܳ�������ˮԡ���Ĺ���
3��Abbe�۹��ǵ�ʹ����ά��
A��У��: Abbe�۹���У������������ⶨ
У���ķ�������Abbe�۹����������ɾ���̨���ϣ����⾵������װ���¶ȼƣ����Ӻó�������ˮԡ����ͨ�����ˮ��һ��Ϊ20���25�棬�����º��ɿ���ť�����������⾵��ʹ�侵�洦��ˮƽλ�ã�����1-2�α�ͪ�ھ����ϣ������⾵����ʹ�ѻӷ����������ߣ��ٴ��⾵����˿�������ֽ�����ò����棬����������ֽ���ر����Ͻ����ֻ��ִ�����ѧ�����
�ñ��۹ⲣ����У��:
���⾵��������1-�������n=1.66���ù⻬�⾵�ϣ��������ճ���ھ����ϣ�ʹ+������ֱ�Ӷ����侵��ת������̶��̣�ʹ�������ڱ�߶���Ϊ����������������ڷ��侵��ʹ���������⾵�飬�Ӳ�����Զ���й۲죬ʹ�ӳ���������ת����ɫɢ��������������ɫɢ������һ����С��˿����ת���澵Ͳ�·��ķ���������ʹ�������ߺ͡�ʮ���ֽ����غϣ�У������������
��������ˮУ����
���⾵����1~2��������ˮ�ھ����ϣ��ؽ��⾵��ת������̶��̣�ʹ�������ڱ�߶�������������ˮ���۹�ȣ�nD20=1.33299��nD25=1.3325�������ڷ��侵��ʹ���������⾵�飬�Ӳ�����Զ���й۲죬ʹ�ӳ����������ڲ�������ʹ�ӳ���������ת����ɫɢ��������������ɫɢ������һ���Ƶ�С��˿���������澵�·��ķ���������ʹ�������ߺ͡�ʮ���ֽ����غϣ�У�������������
B���ⶨ
���������ú��⾵���õιܰѴ���Һ��2~3�ξ��ȵ���ĥɰ�⾵�ϣ�Ҫ��Һ����������ӳ����ؽ��⾵��ת�����侵��ʹ�ӳ�����������ת������̶��̣������Ҿ�Ͳ���ҵ������ֽ���ɫ�������ת����ɫ��������������һ�������ֽ��ߣ�ת������̶��̣��Ƿֽ��߶���ʮ���ֽ�����ϣ������۹��ʡ��ظ�2~3�Ρ�
C����
a. Abbe�۹�����ʹ��ǰ����Ҫ�ñ�ͪϴ�����ò���ֽ����Һ�塣
b. �����������������ˮ��
c. ���ܷ����չ�ֱ�����Դ�ĵط���
d. �ᡢ��ȸ�ʴ��Һ�岻��ʹ��Abbe�۹��ǡ�
e. �۹��Dz���ʱӦ�������ڣ��������������ڿ�����ͨ�����ڡ�
�壮ע������
1������ҺӦ�μ��ʵ���������ٻ�ֲ�������Ӱ��۲⣬�ӷ�Һ��Ӧ���ٲ�����
2�����۹���ʱӦ���к���ˮ�ۣ���֤�ⶨʱ�����¶ȡ����ʵ�����������ޣ��ⶨҺ���۹��ʵ��¶���涨�¶Ȳ�һ�£�ͨ����20���25��Ϊ����������õĽ�������У�����������¶�����1��ʱ��Һ���л�����۹��ʼ���Լ4��10-4����ֵ���л��㡣
����˼����
1���ⶨ�л����۹��ʵ�������ʲô��
2���ٶ�����ɽ��͵��۹���ΪnD30=1.4710����25��ʱ���۹���Ӧ�Ƕ��٣�
ʵ���� �� ��
һ��ʵ��Ŀ��
1���˽�����ԭ�������壬�������������ѡ�÷�����
2��ѧϰʵ���ҳ��÷���IJ���������
��������ԭ��
�����ŵĻ������������һϵ�е��Ƚ��������е㲻ͬ�����ʷ��������Ӱ�����Ч�ʵ�������Ҫ�������������������Ⱥ����ı���Ч����
����ʵ��������ҩƷ
��ԡ�����������������������������ܡ�������β�ܡ�Բ����ƿ���¶ȼơ���ͪ
�ġ�ʵ��װ��
a. b. c.
������
a. ���η������� ����װ��
b. ά�ϣ�Vigreux��������
c. ��ķ����Hempel��������
�塢ʵ�鲽��
1. ������װ��ͼ��װ������
2. ������100mL����ƿΪ���ܹܣ��ֱ�ע��A��B��C��
3. ��250mLԲ����ƿ�ڷ���100mL��ͪ��50mLˮ��������ӣ�������������������ʼ�������ȣ������Ƽ����ٶȣ�ʹ���Һ��ÿ��1~2�ε��ٶ��������������Һ�ռ����Թ�A��ע���¼�����¶ȼ�������A�����Һ���������������¼ÿ����1mL���Һʱ���¶ȼ��������
�¶ȴ�62�滻��ƿB���ܣ�98������ƿC���ܣ�ֱ��������ƿ�в�ҺΪ1~2mL��ֹͣ���ȡ�
��A��56~62�棬 B�� 62~98�� C�� 98~100�棩
��¼������ֵ����������������Һ��������ƿʱ��������¼����Һ������������¶�Ϊ�����꣬���Һ���Ϊ�����꣬��ʵ��������¶�-������ߣ����۷���Ч�ʡ�
4. ��ͪ-ˮ���������ȽϷ���Ч�ʡ�
������������
1. �����������ԭ����װ��������Щ��ͬ����������ַе�ܽӽ���Һ����ɵĻ�����ܷ��÷������ᴿ�أ�
2. ����ѷ����������¶ȼƵ�ˮ������λ�ò���Щ������Ϊʲô��
ʵ���� ˮ��������
һ��ʵ��Ŀ��
1���˽�ˮ���������ԭ����Ӧ�÷�Χ��
2������ˮ�������������װ��Ͳ������ܡ�
����ʵ��ԭ��
�Ѳ��ܻ�������ˮ������һ���ӷ��Ե��л��������ˮ��ϣ�ͨ��ˮ������ʹ�л�������ˮ������������IJ�����ˮ��������
���ֲ����ܵĻӷ������ʻ����һ��ÿһ��ֶ������������������ţ����Ǹ��Եķ�ѹֻ�ڸ��Դ����ʵı�������ѹ�йأ���PA= PA ��PB = PB �������ֵ�Ħ�������أ�����ѹΪ����ѹ֮�ͣ���
P�� = PA + PB = PA + PB
�ɴ˿ɼ��������ķе㽫����һ��ֵķе㶼�͡��ڳ�ѹ����ˮ��������ˮ����Ϊ���е�һ�࣬���ڵ���100�������½��߷е������ˮһ����������
�л������ˮ�������������������
1. ���ܻ�������ˮ��
2. �ڷ����²���ˮ������ѧ��Ӧ��
3. 100�����ұ�����һ��������ѹ��һ�㲻С��1.33kPa��
����������ҩƷ
������ˮ������������������ƿ��������ͷ���������ܡ���������
ҩƷ���屽
�ġ�ʵ��װ��ͼ
�塢ʵ�鲽��
��ˮ��������ƿ�У�����Լռ����3/4����ˮ�����������װ�ò�©����������飿��������T�ιܵ������У����ȵ����ڡ����д���ˮ����������T�ιܵ�֧�ܳ��ʱ���������������У�ˮ������������֣���ʼ��������������У�������ˮ������������ʹ��ƿ��Һ�������ӣ�����������ƿ�ݻ���ʿ/3ʱ������ˮ���������ٶȲ���ʱ�������ָ�ʯ��������֮��Ҫע��ƿ�ڱ�����������������ң���Ӧ���ȣ����ⷢ�����⡣�����ٶ�Ϊ2~3��/s��
����ʵ��ע���
1. ����ϵͳ�й�·�ܶ���̡�
2. ʵ�������Ӧʱ��ע�ⰲȫ���е�ˮλ�Ƿ�������
3. ˮ������������Ӧ�ŷ�ʯ��
4. ʹ�þƾ���ƺ͵�¯Ӧע�ⰲȫ��
���Һ��ɵļ��㣺
����������ֵ��������巽�̷ֱ��ʾΪ��PAVA=nART PBVB=nBRT
����ʽ��ȵõ���ʽ��
��ˮ�������������£�VA =VB���¶���ȣ�����ʽ�ɸ�дΪ
��
���屽~ˮ��ϵΪ�����������е�Ϊ95��ʱ��ˮ������ѹΪ85.3kPa���屽Ϊ16.0 kPa��������ʽ�õ���
�˽��˵������Ȼ�ڻ����е����屽������ѹ����ˮ������ѹ�����������屽�ķ�����������ˮ�ķ�����������������Һ���屽������ˮ�࣬��Ҳ��ˮ���������һ���ŵ㡣
ʵ���� �ؽᾧ������
һ��ʵ��Ŀ��
1���˽��ؽᾧ�Ǵ������ƹ����л���������ֶΣ�
2��ͨ��ʵ������������ˮ���л��ܼ�������ܼ��ؽᾧ���������л����ʵĸ������IJ���������
����ʵ��ԭ��
�����л������ܼ��е��ܽ��һ�����¶ȵ����߶����ѹ����л����ܽ����ȵ��ܼ���ʹ֮���ͣ���ȴʱ�����ܽ�Ƚ��ͣ��л����������������塣�����ܼ��Ա��ᴿ���ʼ����ʵ��ܽ�Ȳ�ͬ��ʹ���ᴿ���ʴӹ�������Һ��������������ȫ�����������Һ�У��Ӷ��ﵽ�ᴿ��Ŀ�ġ��ؽᾧֻ�������ʺ�����5%���µĹ����л��������ᴿ���ӷ�Ӧ�ֲ���ֱ���ؽᾧ�Dz����˵ģ������Ȳ�ȡ�������������ᴿ��Ȼ�����ؽᾧ�ᴿ��
����ʵ��������ҩƷ
����©��������ƿ����ȫƿ��ѭ��ˮ�á����䡢�ջ���ֽ���۵�ʽ��ֽ��
�ġ�ʵ�鲽��
1��ѡ�����˵��ܼ�
��ѡ���ܼ�ʱӦ���ݡ��������ܡ�ԭ���������������ڽṹ�������Ƶ��ܼ��С����ɲ����йص������ֲᣬ�˽�ij�������ڸ����ܼ��в�ͬ�¶ȵ��ܽ�ȡ�Ҳ��ͨ��ʵ����ȷ����������ܽ�ȡ�
2�������ؽᾧ�����Ƴ��ȵı�����Һ
���ȱ߽��裬���������ܼ���������ȫ�ܽ���ٶ��15�����ҵ��ܼ����в����ٶ���ܼ�������������������塣������ɫ������Һ����������̿����ɫ��������Ϊ��������1��5���������5min��
3�����ȹ��˳�ȥ����������
����Һ�ز�������ջ���ֽ����ˮ©��������ʱ�ٶ�Ҫ�졣
4���ᾧ
��Һ������ȴ�������ᾧ������С��
5������
������ȴ�����ľ��壬ֱ���顰�ɡ�Ϊֹ��ע��ͣ��ʱ��Ҫ�ȴſշ�����ͨ����������ͣ�ã��ɱ������
6���ᾧ��ϴ�Ӻ���
���ܼ���ϴ�ᾧ�ٳ��ˣ���ȥ���ŵ�ĸҺ�����˺�ϴ�Ӻ�Ľᾧ�������������������ܼ�������������ʵ��ķ������и��
�塢˼����
1�� �ؽᾧʱ���ܼ�������Ϊʲô���ܹ���̫�࣬Ҳ���ܹ��٣���ȷ��Ӧ����Σ�
2�� �û���̿��ɫΪʲ��Ҫ������������ȫ�ܽ��ż��룿Ϊʲô��������Һ����ʱ���룿
3�� ʹ�ò���©������ʱ�������ֽ����©���ɿ���ʱ����ʲô���ã�
4�� ֹͣ����ǰ���粻�Ȱγ���Ƥ�ܾ�סˮ�����ã�����ʲô���������
ʵ��� �������뱡�����
һ��ʵ��Ŀ��
1���˽�ɫ�������ᴿ�л�������Ļ���ԭ����Ӧ�á�
2����������������������IJ���������
����ʵ��ԭ��
ɫ����Chromatography�����ɫ�㷨���������ȡ�
ɫ���Ƿ��롢�����ͼ����л����������Ҫ����֮һ��ɫ���Ļ���ԭ�������û����������ijһ�����е��������ܽ����ܣ����䣩�IJ�ͬ���������ԵIJ��죬ʹ��������Һ�����������ʽ��з�����������������ã��Ӷ�ʹ����ַ��롣
ɫ�����л���ѧ�е�Ӧ����Ҫ�������¼�����:
1���������� һЩ�ṹ���ơ���������Ҳ���ƵĻ�������ɵĻ���һ��Ӧ�û�ѧ������������ѣ���Ӧ��ɫ�����룬��ʱ�ɵõ�����Ľ����
2�������ᴿ������ �л��������к��������ṹ���Ƶ����ʣ����׳�ȥ��������ɫ�������Գ�ȥ���ʣ��õ���Ʒ��
3������������ ��������ȫһ�µ����������Ļ������ڱ���ɫ��ֽɫ���ж�����һ�����ƶ����룬�Ʊ���ֵ��Rfֵ������������ɫ�����Լ���������Ĵ��Ȼ�ȷ�������������ƵĻ������Ƿ�Ϊͬһ���ʡ���Ӱ�����ֵ�����غܶ࣬�籡��ĺ�ȣ������������Ĵ�С������ԣ����Եȼ�������¶Ⱥ�չ�������ȡ���ɡ��ӷ��Եȡ����ԣ�Ҫ������ֵı���ֵ�ͱȽ����ѡ�Ϊ�ˣ��ڲⶨijһ����ʱ���������֪��Ʒ���ж��ա�
4���۲�һЩ��ѧ��Ӧ�Ƿ���� �������ñ���ɫ��ֽɫ�۲�ԭ��ɫ�������ʧ����֤����Ӧ������
����ɫ����Ҫ�������������轺��Ϊ����������һЩ��������Һ�����������ı����ϣ��������ܼ�ϴ�ѻ�չ�������ò�ͬ�������ܵ��������IJ�ͬ�������ã����������ܼ��в�ͬ���ܽ�ȣ�Ҳ�������ò�ͬ���������������Ϻ���Һ֮��ֲ�����IJ�ͬ���õ����롣����ɫ����ɲ�����ɫ�ͱ���ɫ�����ַ�ʽ��
��ɫ�׳��õ�������ɫ�ͷ���ɫ�����֡�����ɫ�׳����������轺Ϊ������������ɫ���Թ轺������������ά��Ϊ֧�ּ��������սϴ�����Һ����Ϊ�̶��ࡣ������ɫ��ͨ���ڲ����������������ܴ���Ķ���Ի��״��������������������Ļ������Һ����������ʱ�����ֳɷ�ͬʱ�������������϶ˡ���ϴ�Ѽ�����ʱ�����ڲ�ͬ����������������ͬ������ϴ�ѵ��ٶ�Ҳ��ͬ�������γ��˲�ͬ��Σ����������������϶��°�����������������С�ֱ��γ�����ɫ���������ܼ�ϴ��ʱ���Ѿ��ֿ������ʿ��Դ����Ϸֱ�ϴ���ռ����������ɣ�������ɫ���ָ�������ܼ�����ɫ���е�������ȡ����
����ɫ���ֽб����������ɫ���е�һ�֣��ǿ��ٷ���Ͷ��Է����������ʵ�һ�ֺ���Ҫ��ʵ�鼼��������~Һ����ɫ�ף����汸����ɫ��ֽɫ���ŵ㣬һ����������������Ʒ���������ˣ�����0.01�ˣ��ķ��룻��һ���������������ʱ����������Ӻ�Ӵ��ֿ�����������Ʒ���˷��ر������ڻӷ��Խ�С��ϸ��¶������仯������������ɫ���������ʡ����⣬����ɫ���������������л���Ӧ��������ɫ��֮ǰ��һ�֡�Ԥ�ԡ���
������������ѵ����
��ȷװ������ͼ2-23�����ư塢���������������
��1�� ��2�� ��3��
������ڲ�ͬ�IJ�������չ���ķ�ʽ
���������衿
����ɫ��
1���������Ʊ���ʪ����Ʊ���
������Ʊ��ĺû�ֱ��Ӱ��ɫ�Ľ��������Ӧ���������Һ��Ҫ�̶���������չ��ʱǰ�ز��룬ɫ���Ҳ�����ظ������ձ��з���2g�轺������5~6mL����ˮ�����ɺ�״�������ƺõĽ�����ע����������ز�Ƭ�ϣ������������������ҡ�Σ�ʹ��������ƽ���������������ɺ���л��Ҳ�������۵Ĺ轺���и�����˴�С���á���ʵ���ô˷��Ʊ������5Ƭ��������Ϊ�轺G����0.5%���ȼ���ά����ˮ��Һ���ɽ��ϡ�
2������
����Ǧ���ھౡ���һ��1cm�����Ữһ������Ϊ��ʼ�ߣ�Ȼ����ëϸ����ȡ��Ʒ������ʼ����С�ĵ������ߵ�ֱ��һ�㲻����2mm��������Ʒ��Һ̫ϡ�����ظ���������Ӧ��ǰ�ε������ܼ��ӷ������µ������Է�������������β����ɢ������Ӱ�����Ч��������ͬһ���ϵ㼸��������������ӦΪ1cm���ϡ�����Ҫ�ᣬ���ɴ��Ʊ��㡣
3��չ��
����ɫ��չ������Ҫ���ܱ������н��С�Ϊʹ�ܼ�����Ѹ�ٴﵽƽ�⣬����չ�����ڳ�һ��ֽ���ڲ������м�����õ�չ���ܼ���ʹ��߶Ȳ�����1cm������õı����С�ķ���������У�����һ�˳��£�����չ�����С��Ǻ�ƿ�ǣ��۲�չ����ǰ��������һ���߶�ʱȡ���������ڰ��ϱ���չ����ǰ��λ�á����ɣ��۲�ߵ�λ�ã�����Rfֵ��
4����ɫ
��������ֽɫ����ɫ���������ڱ���ɫ�ס�����ɫ����ʹ�ø�ʴ�Ե���ɫ����Ũ���ᡢŨ�����Ũ����ȡ����ں���ӫ�������п�ӡ�����п��ӫ��ƣ��ı������������¹۲죬չ������л�������������ӫ�ⱳ���ϳ���ɫ�ߵ㡣Ҳ����±�ذߵ����鷨��ʹ����ɫ�װߵ���ɫ����ʵ����Ʒ����������ɫ��������ӫ����¹۲졣
��ɫ��
1��װ����ʪ����
������ȡ������֬���ڸɾ���ɫ�����ײ��������������رջ����������е����ܼ���ԼΪ���ߵ�3/4����ͨ��һ����IJ���©����������ɫ���������������������������������ٶ�Ϊ1��/�룻������Ƥ�������ô�ɫ�����²���ʹ��װ���ܣ���װ����3/4ʱ�����������һƬСԲ��ֽ����֬�ޡ�����ʱһֱ������������ע�ⲻ��ʹҺ��������������ϲ㡣
2������
���ܼ�Һ��պ�������ֽ��ʱ�����������ڼ����������Ʒ��Һ��������Һ�����ӽ���ֽ��ʱ�������������ܼ�ϴ�¹ܱڵ���ɫ���ʣ��������2~3�Σ�ֱ��ϴ��Ϊֹ��
3��ϴ��
������õ��ܼ���ѧ���Լ����ƣ�ϴ�ѣ����������ٶȡ��������̶�Ӧ��ϴ�Ѽ�����������������С��ɫ�����������ƶ������Խϴ�����������϶ˣ��γɲ�ͬ��ɫ�����۲�ɫ���ij��֣�������ƿ�ռ�ϴ��Һ��
�ġ�ʵ��ؼ���ע�����
1���������ʱ���������Ʊ�Ҫ���ȣ�����ƽ����ࡣ
2������ʱ����������1~1.5cm������ֱ��Ӧ������2mm��
3��������ʱ������Ҫ���ܽ�ʵ�������ݡ�
�塢��Ҫ�Լ�����Ʒ������������������ֵ��
�� ��
������
��״
�۹���
����
�۵��
��
�ܽ�ȣ���/100mL�ܼ�
ˮ
��
��
ż����
182.2
1.2000
6 8
293
4.220
12 20
����������
138.13
112~115
0.11
7.120
7.920
ӫ���
332.3
320
�������ʸ�Ҫ
1��Ϊʲô���Դ�����Ҫ�ü��Խϴ���ܼ�ϴ�ѣ�
2���������������ݻ�װ����ȣ������������ʲô���Ľ������α��⣿
3���������Rfֵ�����������
�ߡ���Ҫ�Լ����������
1%ż������1%������������ӫ���(95%�Ҵ�, 1mg/1mL)���μ�����������:���� 9:1
ʵ��� ��ȡ��ϴ��
һ��ʵ��Ŀ��
1���˽���ȡ����Ļ���ԭ�����˽��黯�����黯����;
2���������շ�Һ©���Ļ���������ʹ��ע�����
����ʵ��ԭ��
��ȡ���������������ֲ����ܣ����ܣ��ܼ����ܽ�Ȼ����ȵIJ�ͬ���ﵽ���롣 ��ȡ
���л���ѧʵ����������ȡ���л�������ij��÷���֮һ��Ӧ����ȡ���Դӹ����Һ����������ȡ���������ʣ�Ҳ��������ϴȥ����������������ʡ�ͨ����ǰ��Ϊ����ȡ������ȡ������Ϊ��ϴ�ӡ���
����ʵ��������ҩƷ
Һ����ȡ��ͨ���������Ƿ�Һ©����һ��ѡ���ݻ��ϱ���ȡҺ��1~2���ķ�Һ©����
�ġ�ʵ�鲽��
�ڷ�Һ©��������Ϳ����֬��������ת��Ȧ��ʹ��֬���ȷֲ�������С��ƤȦ��ס����β����С�ۣ���ֹ�������ѡ��غû�����װ�����ȡ�����ȡ�ܼ����������ӣ���������������ʳָĩ�ڽ�©���϶˲�����ס�����ô�Ĵָ��ʳָ����ָ��ס©���������ֵ�ʳָ����ָ�����ڻ����ı��ϣ�����������ҡ��Һ©����ʹ����֮���ֽӴ����������ȡЧ�ʡ�ÿ��ҡ���κ�Ҫ��©��β��������б�������˴��������������Խ��©���е�ѹ��������ظ�������ʱֻ�к�Сѹ�����پ�����ҡ2��3min�����ã���������ȫ�ֿ�������IJ������ٽ����������������²�Һ���Ի����ų�����ʱ���������ܳ���һЩ��״��ҲӦͬʱ��ȥ��Ȼ���ϲ�Һ��ӷ�Һ©���Ͽڵ�����ȴ����Ҳ�ӻ����ų������ⱻ������©�����ϵ���һ��Һ����մ�ۡ�
�塢˼����
�����黯�������ķ���ͨ������Щ��
(1) �ϳ�ʱ�侲�ã�
(2) ��������Զ������黯���ɼ����������ƻ�����ù��˷�����ȥ��
(3) �������������ܼ���ˮ���л��ܼ����ܲ��ֻ��ܶ������黯���ɼ�����������ʣ����Ȼ��Ƶȣ��������������ü����ƻ������⣬����ʳ�Σ�������ˮ��ı��أ������������������Сʱ�ķ��룻
(4) �������ƻ���״Һ����μӼ����Ҵ����ǻ������͵��Խ��ͱ���������
����ʵ����ϸ��������ʲô�����ȡʲô��ʩ��
��ѧ��ȡ
��ѧ��ȡ��������ȡ���뱻��ȡ����ѧ��Ӧ��Ҳ�dz��õķ��뷽��֮һ����Ҫ����ϴ�ӻ�����������������ǰ��ķ�����ȡ��ͬ�����磬���ü�����ȡ�����л�������ȡ���л��ᣬ��ϡ����Դӻ��������ȡ���л��������ʻ����ڳ�ȥ�������ʣ���Ũ����ӱ�����
�г�ȥ������������±�����г�ȥ�����ѵȡ�
Һ~����ȡ
�Թ�������ȡ�����ͨ�����ó��ڽ����������֬����ȡ����ǰ���ǿ��ܼ����ڵĽ����ܽ�������������е���Ҫ�ɷֽ�������Ч�ʵͣ��ܼ�����
֬����ȡ���������ܼ������ͺ���ԭ�����ǹ�������ÿһ�ζ��ܱ������ܼ�����ȡ�����Ч�ʽϸߣ�Ϊ����Һ����ܵ��������ȡǰӦ�Ƚ�������ϸ������ֽ�װ���������ȡ���У���ȡ���¶˽�ʢ����ȡ������ƿ���϶˽������ܣ����ܼ�����ʱ�������������ܼ�������ȡ���У���Һ�泬���������϶˺�����������ƿ�������ȡ�������ܼ��IJ������ʡ������������ܼ������ͺ������ã��ǹ����еĿ������ʸ�������ƿ�У���ȡҺŨ�������ù����һ���ᴿ��
ʵ��ʮ ��ѹ����
һ��ʵ��Ŀ��
1���˽��ѹ�����ԭ����Ӧ�÷�Χ��
2�� ��ʶ��ѹ�������Ҫ�����豸��
3�� ���ռ�ѹ���������İ�װ�Ͳ���������
����ʵ��ԭ��
Һ��ķе���ָ��������ѹ�������ѹ��ʱ���¶ȣ����Һ��ķе��������ѹ���ı仯���仯�ģ������������ձý���ϵͳ��ѹ�����Ϳ��Խ���Һ��ķе㣬����Ǽ�ѹ����������������ݡ�
��ѹ�����Ƿ��롢�ᴿ�л�������ij��÷���֮һ�����ر���������Щ�ڳ�ѹ����ʱδ��е㼴�����ȷֽ⡢������ۺϵ����ʡ�
����ʵ��������ҩƷ
��ѹ����װ����Ҫ������������ѹ������ȫ�����Ͳ�ѹ�IJ�����ɡ�
�ġ�ʵ�鲽����˵��
1��������Һ���������еͷе����ʣ�ͨ���Ƚ��г�ѹ��������ˮ�ü�ѹ����������ͱü�ѹ����
2����װ��װ�ú�����ʹ��ϵ�������ͨ�������ͱó������رն�ͨ��������ȫ�رգ�ע��۲�ƿ�ڵĹ���������緢�ֹ���̫���ң��г���Σ�գ���������ͨ��������Щ����ѹ�����Ϲ۲���ϵ��ѹ��Ӧ�ܷ���Ҫ��Ȼ��С��������ͨ������ͬʱע��۲�ѹ�����ϵĶ�����������ϵ��ѹ������ֵ�����ݷе���ѹ����ϵ����
3����ϵͳ��ֳ�պ�ͨ����ˮ��������ԡ��������һ����ѹ����ʼ����Ӧ����ע�����������������ϵ��ѹ��������¼ѹ������Ӧ�ķе�ֵ������Ҫ���ռ���ͬ��֡�
4��������ϣ���ȥ��Դ���������������У���ֹ����������������ͨ������ƽ������ѹ����ʹ��ѹ�Ƶ�ˮ���������ػظ�ԭ״��������̫�죬ˮ�����ܿ��������г��Ʋ�ѹ�ƵĿ��ܣ���Ȼ��ر��ͱú���ȴˮ��
�塢˼����
1��������������²��ü�ѹ����
2��ʹ���ͱü�ѹʱ��ʵ����Щ���պͱ���װ�ã���������ʲô��
3���ڽ��м�ѹ����ʱ��Ϊʲô��������ԡ���ȣ���������ֱ�ӻ���ȣ�Ϊʲô���м�ѹ����ʱ���ȳ������ܼ��ȣ�
4������ѹ������Ҫ�Ļ������Ӧ���ֹͣ��ѹ����Ϊʲô��
ʵ��ʮһ ����ϩ���Ʊ�
һ��ʵ��Ŀ��
1����Ϥ������������µķ�Ӧ���������ջ���ϩ���Ʊ�������
2�����̷�Һ©����ʹ�ã���ϰ���������
����ʵ��ԭ��
�����Լ�
�����������ᣨ85%����ʳ�Σ���ˮ�Ȼ��ƣ�5%̼������Һ��
�ġ�ʵ�鲽��
ʵ��װ�����£�
��50���������Բ����ƿ�У�����10.4mL��������5mL85%���ᣨ��2mLH2SO4���ͽ�����ӡ���ƿ��װһ�̵ķ�����������װ�ã����������ܣ�����ƿ�������������ñ�ˮ��ȴ��
��������������������ƿ����ԡ����С���������ȣ����Ƽ����ٶ�ʹ�������϶˵��¶Ȳ�Ҫ����90�棬��ҺΪ��ˮ�Ļ�������ƿ��ֻʣ�º������IJ����������������ʱ������ֹͣ����ȫ������ʱ��Լ��1h��
������Һ�þ��Σ�1g���ң����ͣ�Ȼ�����3~4mL 5%̼������Һ�к������ᡣ����Һ�嵹��С��Һ©���У���ҡ���÷ֲ㡣���²�ˮ��Һ��©���¶˻����ų����ϲ�Ĵֲ�����©�����Ͽڵ�������С��ƿ�У�����1~2����ˮ�Ȼ��Ƹ��
�������IJ���������������ƿ�У���ˮԡ���������ռ�80~85��������һ�ѳ��صĸ���С��ƿ�С�
�塢ע������
1���������ڳ�������ճ��״Һ�壬���������Ͳ��ȡʱӦע��ת���е���ʧ��
2��ˮ��Ӧ�����ܷ�����ȫ������������ˮ�Ȼ��Ƶ�������ʹ�������ر������������������ʧ����������ˮ�Ȼ��Ƹ�����ʺϣ��������ɳ�ȥ������������
3���������Ѹ���IJ���ʱ����������������Ӧ��ָ��
����˼����
1�����з������ʱӦע��ʲô��
�𣺢�. ������װ��ʱӦʹ�����������������洹ֱ���Ա�֤��������������Һ��������������������г�ֵ��Ƚ������ʽ�������߷���Ч������. ���ݷ���Һ��ķе㷶Χ��ѡ�ú��ʵ���ԡ���ȣ���Ҫ��ʯ��������ֱ�ӻ���ȡ���С�������ԡ���Ա�ʹԡ�»��������ȵ������� ��. Һ�忪ʼ���ڣ����������������ʱ��Ҫע�����ԡ�£�ʹ����������������
���ط������������������µͻ�Һ��е�ϸߣ�Ӧ����������ʯ���������������������Լ���������������ʧ����. ��������������������������ʼ��Һ�����ʱ��Ӧ����ע�����ԡ�£��������Һ���ٶ�Ϊÿ2��3��/s����������ٶ�̫�죬��Ʒ�����½������ٶ�̫�������������������ʱ��ʱ��������¶Ȳ�������. ����ʵ��涨��Ҫ�ֶμ�ȡ��ݣ�ʵ�����ʱ������������ݡ�
2���ڻ���ϩ�Ʊ�ʵ���У�ΪʲôҪ���Ʒ��������¶Ȳ�����73�棿
����Ϊ��Ӧ�л���ϩ��ˮ�γɹ��л����(�е�70.8�棬��ˮ10 %)���������뻷��ϩ�γɹ��л����(�е�64.9�棬��������30.5 %)����������ˮ�γɹ��л����(�е�97.8�棬��ˮ80 %)����ˣ��ڼ���ʱ�¶Ȳ��ɹ��ߣ������ٶȲ����죬�Լ���δ��Ӧ�Ļ�������������
3������ϩ���Ʊ������У�������ʵ�����̫�ͣ��Է�����Ҫ����Щ���������������ʧ��
��(1)��������ճ�Ƚϴ��������µ�ʱ����Ͳ�ڵĻ��������ѵ�����Ӱ����ʡ�(2)����ͻ�������ϲ���������ʱ����̼����(3)��Ӧ�¶ȹ��ߡ�����ٶȹ��죬ʹδ��Ӧ�Ļ���������ˮ�γɹ��л�������ﻷ��ϩ��ˮ�γɹ��л�����Ӱ����ʡ�(4)�����������������ʱ����̣���ʹ���������ǰ��������Ӱ����ʡ�
�ߡ���������
1���ڴ��ƵĻ���ϩ�У����뾫��ʹˮ�㱥�͵�Ŀ�ĺ��ڣ�
2����������ֹǰ�����ֵ����������ʲô��
ʵ��ʮ�� ���ѵ��Ʊ�
һ��ʵ��Ŀ��
1. ����ʵ�����Ʊ����ѵ�ԭ���ͷ�����
2. �������յе���ȼҺ��IJ���Ҫ�㡣
����ʵ��ԭ��
����Ӧ��
�ܷ�Ӧ��
����Ӧ��
����ʵ��������ҩƷ
�����ס�ˮԡ����������ƿ����Һ©�����¶ȼơ������ܡ����������۹��ǡ��Ҵ���ŨH2SO4��5%NaOH������NaCl��CaCl2�����ͣ�����ˮCaCl2
�ġ�ʵ�鲽��
1. ���ѵ��Ʊ�
�ڸ����������ƿ�м���12mL95%�Ҵ�����������12mLŨH2SO4��Ͼ��ȡ�
�� ����Һ©���м���25mL95%�Ҵ���
�� ��ͼ���Ӻ�װ�á�
�� �õ������ȣ�ʹ��Ӧ�¶ȱȽ�Ѹ������140�档��ʼ�ɵ�Һ©�������μ��Ҵ���
�� ���Ƶ����ٶ������Һ�ٶȴ�����ȣ�1��/s����
�� ά�ַ�Ӧ�¶���135~145����30~45min���꣬�ټ�������10min��ֱ���¶�����160�棬ֹͣ��Ӧ��
2. ���ѵľ���
�� �����Һת����Һ©���У�������8mL5%NaOH��8mL����NaClϴ�ӣ������8mL����CaCl2ϴ��2�Ρ�
�� �ֳ��Ѳ㣬����ˮCaCl2���
�� �ֳ��ѣ������ռ�33~38����֡�
�� ������ʡ�
�塢ע������
1��������Ũ���ᣬ�ӱ�ҡ����ȴ����ֹ�Ҵ�������
2��װ��Ҫ���ܣ���Ӧ���Ҫ��ͣ������ȴ���ٲ��½���������ֹ����ӷ���
3�����ƺ÷�Ӧ�¶ȼ��μ��Ҵ����ٶ�1d/s��
4��ϴ��ʱע��˳������������
5���־�ˮ������ˮ�Ȼ��Ƹ���Լ20~30min��
6�����ý��Ȼ��ƴ�����ƿ������ˮԡ������������
7����Ʒ��������ͷе㡣
������������
1. ��ʵ���У��ѻ��ڴ��������������һһ��ȥ������Щ��ʩ��
2. ��Ӧ�¶ȹ�����ͶԷ�Ӧ��ʲôӰ�죿
3. ���ݴ���Ӧ�й��̡�
ʵ��ʮ�� 1-�嶡����Ʊ�
һ��ʵ��Ŀ��
1��ѧϰ���廯�ơ�Ũ������������Ʊ�1-�嶡���ԭ���ͷ�����
2���������մ��������к�����װ�õĻ����Ȼ�����������һ������Һ���������ᴿ�ķ�����
����ʵ��ԭ��
����Ӧ��
���ܵĸ���Ӧ��
����ʵ��������ҩƷ
���������ܡ�Բ����ƿ����������װ�á�ˮԡ����װ�á�����װ�á�β������װ�á���Һ©����ŨH2SO4����������NaBr��NaOH����ˮCaCl2��NaHCO3
�ġ�ʵ�鲽��
1. ��100mLԲ����ƿ�У�����10mLˮ�����������ӣ���������12mL(0.22mol)ŨH2SO4�������������¡�
2. ����������7.5mL(0.08mol)����Ϻ����10g��ϸ���廯�ƣ������
3. ��װ��ͼװ�ã���5%NaOH�����ռ���
4. �������������������Ȼ���0.5h����ȴ��������װ�ã���ʯ�����ϼ������������嶡�顣
5. ���Һת���Һ©������10mLˮϴ�ӣ�С�ĵؽ���Ʒת�뵽��һ����ķ�Һ©���У���5mLŨH2SO4ϴ�ӡ�
6. ������ȥ����㣬�л������ηֱ���ˮ������NaHCO3��ˮ��10mLϴ�ӡ�
7. �����������С������ƿ�У�������ˮCaCl2�����϶ҡ������Һ������
8. �����ռ�99~103����֡�
�塢��������
1. ��ʵ���У�Ũ����������ã���������Ũ�ȶ�ʵ���к�Ӱ�죿
2. ��Ӧ��Ĵֲ�Ʒ�к�����Щ���ʣ���������α���ȥ�ģ�
3. Ϊʲô�ñ���̼������ˮ��Һϴ����ǰ��Ҫ����ˮϴ�ӣ�
ʵ��ʮ�� �����������Ʊ�
һ��ʵ��Ŀ��
1������л���ϳ�����һ��ԭ����������
2����һ����������Һ©����ʹ�õȻ���������
����ʵ��ԭ��
����������ҩƷ
���������ܡ�Բ����ƿ����Һ©��������װ�á���ˮ�Ҵ��������ᡢŨH2SO4������Na2CO3������NaCl������CaCl2����ˮMgSO4��
�ġ�ʵ�鲽��
1����50mLԲ����ƿ�м���9mL(0.2mol)��ˮ�Ҵ���6mL(0.10mol)�����ᣬ��С�ļ���2.5mLŨ���ᣬ���Ⱥ�����������ӣ�װ�������ܡ�
2��С����ȷ�Ӧƿ�����ֻ���30min����ƿ�ڷ�Ӧ����ȴ������װ�øij�����װ�ã�����ƿ����ˮ��ȴ�������������ɵ�����������ֱ�����Һ���ԼΪ��Ӧ���������1/2Ϊֹ��
3�������Һ���������뱥��Na2CO3������������CO2�������Ϊֹ��
4�������Һת���Һ©������ȥˮ��Һ���л�����5mL����NaClϴ�ӣ�����5mLCaCl2ϴ�ӣ������ˮϴ��һ�Σ���ȥ�²�Һ�塣
5���л��㵹�����������ƿ�У�����ˮMgSO4���
6����ѹ�����ռ�72 ~78����֡�
��2��
�塢˼����
1��������Ӧ��ʲô�ص㣿��ʵ������δ���������ʹ������Ӧ�����������﷽����У�
2����ʵ�������ô�������������Ƿ���ʣ�Ϊʲô��
3�� �����Ĵ�������������Ҫ����Щ���ʣ���γ�ȥ��
ʵ��ʮ�� �����������Ʊ�
һ��ʵ��Ŀ��
1. ���ձ�����������Ӧ��ԭ����ʵ�������
2. ��һ�������ؽᾧ���ӷ�Ӧϵͳ���ᴿ����ķ�����
�� ��ʵ��ԭ��
����������ҩƷ
Բ����ƿ�����������¶ȼơ���ԡ����װ�á���������װ�á��ؽᾧװ�á������������ᡢZn�ۡ�����̿
�ġ�ʵ�鲽��
1��50mLԲ����ƿ�У�����10mL��������ı���(0.11mol)��15mL������(0.26mol)������п��(Լ0.1g)��
2��װ�Ϸ������������¶ȼƣ�����ͼ��ʾ��
3����������Լ1h��
4����Ӧ���������£����Ƚ���Ӧ�ﵹ��ʢ��250mL��ˮ�ձ��У���ȴ���ˣ�ϴ�Ӵֲ�Ʒ��
5�����ֲ�Ʒ�ؽᾧ�����ˣ����ء�
�塢˼����
1�����ؽᾧʱ��Ҫע����Щ����ſ���ʹ��Ʒ���ʸ㡢�����ã�
2�� ��ӦʱΪʲôҪ�����������϶˵��¶���100��110��֮���أ��¶ȹ�����ʲô���ã�
ʵ��ʮ�� �������Ʊ�
һ��ʵ��Ŀ��
1�� ͨ���������Ʊ�ѧϰ������Perkin��Ӧ�������������
2�� ���̡�����ˮ���������ԭ�����ô��Ͳ�����
3�� ���̲����չ����л���������ᴿ��������ɫ���ؽᾧ��
����ʵ��ԭ��
����ʵ���������Լ�
Բ����ƿ�������ܡ��¶ȼƣ� 250�棩�� ˮ��������װ�á�����ȩ����ˮ̼��ء� ��ˮ̼���ơ���������Ũ���ᡢ������̿
�ġ���������
��250mLԲ����ƿ�м���4.1g��ϸ����ˮ̼��أ� 3.0mL������ı���ȩ(0.03mol)��5 .5mL ������(0.06mol)�����������ӡ�Բ����ƿ���ϻ��������ܣ��������϶˽Ӹ���ܡ���ʯ�����ϼ���ʹ���������ӦҺʼ�ձ�����150��170�棬ʹ��Ӧ����1h(����1h)��
ȡ��Բ����ƿ�������м���50 mLˮ��10.0g̼���ƣ�ҡ����ƿʹ�����ܽ⡣Ȼ�����ˮ��������(��ȥδ��Ӧ��ȫ�ı���ȩ)��ˮ����������������Һ��������Ϊֹ��
ж��ˮ��������װ�ã���Բ����ƿ�м��롫1.0g����̿�����ȷ���2��3min��Ȼ������ȹ��ˡ�����Һת�����ɾ���200mL�ձ��У���������Ũ��������ữ�����Ե����ԣ���Լ��25mLŨ���ᣩ��Ȼ�������ȴ��������ֽᾧ��֮����м�ѹ���ˡ�������������ˮϴ�ӡ���ѹ���ˣ�Ҫ��ˮ�ֳ��׳�ɣ��ڿ��������ɣ�����(�ɵ�2��2 .5g��Ʒ)��
�塢˼�����ע������
1�� ���к��ֽṹ��ȫ�ܹ�����Perkin��Ӧ��
2�� �������ˮ����������ϼ�������������Ҫ����̼����ʹ��Һ�ʼ��Ժ�ſ��Խ���ˮ��������Ϊʲô���ܷ����������ƴ���̼���ƣ�
3�� Perkin��Ӧ�����������볹���������ȡ����ȩ������������Ͳ������������ˮ̼��غ���ˮ�������Ϊ���ϼ������Dz�������ˮ̼���ơ�����ʱ����ǿ�Ȳ���̫������������������Ϊ�˽�ʡʱ�䣬�����ڻ�������֮ǰ��30min��ʼ����֦����ƿʹˮ���ڣ������û�ֱ�Ӽ�����ƿ��������ɫ����ʱһ��ȡ����ƿ������֮���ټ��Ȼ���̿���ȹ���ʱ�����������ȹ��ˣ�����©��Ҫ�����ڷ�ˮ��ȡ��������Ҫ�졣�����ữʱҪ��������Ũ���ᣬһ����Ҫ����̫�죬�����Ʒ����ձ���ɲ�Ʒ��ʧ�������Ҫ�ᾧ���ף���������ˣ�������̫��ˮϴ�Ӳ�Ʒ��
ʵ��ʮ�� �Ӳ�Ҷ����ȡ������
һ��ʵ��Ŀ��
1�� ѧϰ�Ӳ�Ҷ����ȡ������Ļ���ԭ���ͷ���, �˽�����һ�����ʡ�
2��������������ȡ����ȡ�л����ԭ���ͷ�����
3����һ����Ϥ��ȡ�����������Ȼ���������
����Ԥϰ����
1����ȡ
2���������
3����������
4����Ȼ����ķ����ᴿ�ͼ������������֪ʶ
������������
1��ʵ������
��Ҷĩ
��ȡҺ
����ȡҺ
����ȡ��
������
������ȡ
95%���Ҵ�
����
����
1������
2���ռ�
2������(Soxhlet)��ȡ��
����(Soxhlet)��ȡ������ƿ����ȡͲ������������3�������, װ��������ͼ��ʾ��������ȡ���������ܼ��Ļ���������ԭ����˼����1��,ʹ��������ÿ�ζ����������ܼ�����ȡ, �������ܼ�����, ��������ȡʱ��, ���Ч�ʽϸߡ���ȡǰ, Ӧ�Ƚ�����������ϸ, �������ܼ����������˼����2����Ȼ����ϸ�Ĺ�������װ����ֽͲ�ڣ�˼����3��4��, �����ڳ���Ͳ, ��ƿ��ʢ��, �������Ͳ����, ����Ͳ��ʽ��ȡ���϶˽������ܡ��ܼ����ȷ���, �������س���Ͳ���������������, ����ΪҺ��, ������ֽͲ��, ������Ͳ����Ʒ����Һ�泬����������ߴ�ʱ, ������������ƿ, �Ӷ���ȡ�������ܼ��IJ������ʡ���˶���ظ�,��Ҫ��ȡ�����ʸ�������ƿ�ڡ���ȡҺ��Ũ����ȥ�ܼ���, ���ò���, ��Ҫʱ��������������һ��������
˼����1����ʽ��ȡ���Ĺ���ԭ����
˼����2����ʽ��ȡ�����ŵ���ʲô��
˼����3��������ʽ��ȡ����ֽͲ�Ļ���Ҫ����ʲô��
˼����4��ΪʲôҪ���������ʣ���Ҷ����ϸ�ɷ�ĩ��
3������װ��
�ġ�ʵ��ԭ����
�������ֽп��ȼ�, ��һ������� , �����ڲ�Ҷ�����ȡ��ɿɵ�ֲ���С������Ҷ�к��� 1%��5%�Ŀ�����, ͬʱ�����е����ᡢɫ�ء���ά�ص����ʡ�
�������������Ի�����, �������ȷ¡��������Ҵ�����ˮ��, ���������Ѻͱ�(��)����Ʒ��
��235��236��, ���ᾧˮ�Ŀ�����Ϊ��ɫ��״����, ��100 ��ʱʧȥ�ᾧˮ, ����ʼ������120 ��ʱ����������178��ʱѸ��������������һ���ʿɴ�������������ĽṹʽΪ :
������1,3,7-����-2,6-�������ʣ�
��������һ���º͵��˷ܼ�, ���д̼����ࡢ�˷���������������á�
��ȡ������ķ����м�Һ��ȡ����������ȡ����ȡ������ʵ�����Ҵ�Ϊ�ܼ�, ��������ȡ����ȡ , �پ�Ũ�����к͡�����, �õ����ᾧˮ�Ŀ�����
��ҵ�Ͽ�������Ҫ��ͨ���˹��ϳ��Ƶá������д̼����ࡢ�˷ܴ�������������á��ʿ�����Ϊ�������˷�ҩ����Ҳ�Ǹ�����˾ƥ�֣�A.P.C����ҩ������֮һ��
�塢ʵ�鲽�裺
1�� ���������ȡ
��ȡ5g�ɲ�Ҷ, װ����ֽͲ��, ����ѹʵ, ��ֽͲ�Ͽ���һ����֬�ޣ�˼����5��, ���ڳ���Ͳ��, Բ����ƿ�ڼ��� 60��8OmL95% �Ҵ�, �����Ҵ�����, ��������1h, ������Һ�ոպ�����ȥʱ, ����ֹͣ���ȡ�
��������װ������װ��, ���Ȼ��մ��Ҵ���Ȼ����Һ (��Լ10��15mL) ������������, ��ƿ�������Ҵ�ϴ��, ϴ��ҺҲ������������, ���������ɡ�����4g ��ʯ�ҷۣ�˼����6��, �������, �õ�������(100��120V), ��������, ��ȥȫ��ˮ�֣�˼����7������ȴ��, ��ȥմ�ڱ��ϵķ�ĩ, ��������ʱ��Ⱦ���
��һ�Ŵ�������С��Բ����ֽ������������, ȡһֻ��С���ʵIJ���©����������, ©���������ɵ���һ������˼����8����
�õ�����С�ļ���������, ���������¶�, ʹ������������������ͨ����ֽ������©���ڱ���Ϊ����, ������©���ڱں���ֽ�ϡ���ֽ�ϳ��ְ�ɫ�� ״����ʱ, ��ͣ����, ���� 100 ������, �ҿ�©������ֽ, ��ϸ��С���Ѹ�������ֽ��©�����ϵĿ��������������С����������ڵIJ������Խ���, ���·ź���ֽ��©��, �ýϸߵ��¶��ټ�������һ�Ρ���ʱ���¶�Ҳ����̫�ߣ������������ڴ���ð�̣���Ʒ������Ⱦ������ʧ��˼����9�����ϲ������������ռ��Ŀ�����, �ⶨ�۵㡣
2��������ļ���
(1) ��������Լ���ȡ������ᾧ��һ����С�Թ��У� �� 4CmLˮ���ȣ�ʹ�����ܽ⡣��װ��2 ֧�Թ��У�һ֧����1��2 �� 5% ������Һ����¼����˼����10������һ֧�� 1��2 �� 10% ���� ( �� 10% ���� )���ټ��� 1��2 �ε�һ�⻯���Լ�����¼����˼����11����
(2) �������ڱ�����ʣ��Ŀ������У�����30% H2O2 8��10�Σ�����ˮԡ������, ��¼����
��ɫ���ټ�һ��Ũ��ˮ�ڲ�����, �۲첢��¼��ɫ�кα仯��˼����12��?
˼����5��ΪʲôҪ����һ����֬�ޣ�
˼����6����ʯ�ҵ�������ʲô��
˼����7��Ϊʲô�������ˮ�֣�
˼����8������װ���У�ΪʲôҪ���������ϸ��Ǵ���С����ֽ��©����Ϊʲô������
˼����9�����������У�Ϊʲô�����ϸ�����¶ȣ�
˼����10����������������Һ��������ʲô������
˼����11�����������~�⻯���Լ���������ʲô��ɫ�ij�����
˼����12���������������������������õ�ʵ��������ʲô��
�������ڵ�������ע�����
1����ֽͲ��ֱ��Ҫ��С�ڳ���Ͳ���ھ� , ��߶�һ��Ҫ����������, ������Ʒ���ø��ں����ܡ������ֳɵ���ֽͲ, �������������䷽��Ϊ ��ȡ��֬��ֽһ��, ����ԲͲ״ ( ��ֱ����С�ڳ���Ͳ�ھ�), �ײ���������(��Ҫʱ����������), װ����Ʒ, �Ͽڸ���֬��, �Ա�֤����Һ���ȵؽ�����ȡ�
2����ȡ�����У���ʯ�����кͼ���ˮ���� ��
3����ʽ��ȡ���ĺ����ܼ����۶ϣ�װ��װ�ú�ȡ��ʱ�����ر�С�ġ���ȡʱ��������ƿ��������ˮ��, ������ʼʱ, ������һЩ����, ��Ⱦ����Ͳ�Ʒ��
4���������ϸ��Ǵ���С����ֽ��Ϊ�˱����������Ŀ������������������, ֽ�ϵ�С��Ӧ��֤����ͨ����©����������Ϊ��ֹ�����������ݳ���
5�������������б���ʼ���ϸ���Ƽ����¶�, �¶�̫��, �����±��������ֽ̿��, һЩ��ɫ����Ҳ�ᱻ������, Ӱ���Ʒ���ʺ���������������ʱ, �����¶���Ӧ�ϸ���ơ�
�ߡ��������ۣ�
���������������
���������ͨ���ⶨ�۵㼰�������Լ��𡣴��⣬������ͨ���Ʊ�������ˮ�������������һ��ȷ֤����������Ϊ�����ˮ������������ˮ�����Σ����ε��۵�Ϊ137�档
������ ˮ���� ������ˮ������
������ˮ��������������Ʊ����������Թ��м���50mg������37ˮ�����4�ױ�����ˮԡ�ϼ���ҡ��ʹ���ܽ⣬Ȼ�����Լ1ʯ���ѣ�60~90�����ڱ�ԡ����ȴ�ᾧ�������������������ò�������ε�Ħ���ܱڡ��ò�����©�������ռ�����ⶨ�۵㡣���ε��۵�137�档
�ˡ�������
1�� ����������ȡ������ȡԭ��������һ��Ľ�����ȡ��ȣ�����Щ�ŵ㣿
��������ȡ���������ܼ��Ļ���������ԭ����ʹ��������ÿ�ζ����������ܼ�����ȡ, �������ܼ�����, ��������ȡʱ��, ���Ч�ʽϸߡ�
2�� ��ʽ��ȡ�����ļ�������ɣ�
������(Soxhlet)��ȡ������ƿ����ȡͲ�����������ܵ���������ɡ�
3�� ��ʵ�������������ʱ��Ӧע��ʲô��
������ȡ������ֵ�����£�����������ʵ��ɰܵĹؼ������������У�ʼ�ն�����С���Ӽ��ȡ����¶�̫�ߣ���ʹ����ơ�ע���¶ȼ�Ӧ���ں��ʵ�λ�ã�ʹ��ȷ��ӳ���������¶ȡ�����ɰԡ��Ҳ�����ü�����ԡ��������������������ײ����뿪ʯ�������м��ȣ����ڸ��������¶ȼ�ָʾ�����¶ȡ�
�š�˼�����
˼����1��������ȡ���������ܼ��Ļ���������ԭ����
˼����2��ʹ��������ÿ�ζ����������ܼ�����ȡ, �������ܼ�����, ��������ȡʱ��, ���Ч�ʽϸߡ���ȡǰ, Ӧ�Ƚ�����������ϸ, �������ܼ����������
˼����3����ֽͲ��ֱ��Ҫ��С�ڳ���Ͳ���ھ� , ��߶�һ��Ҫ����������, ������Ʒ���ø��ں����ܡ���Ԫ�ֳɵ���ֽͲ, �������������䷽��Ϊ ��ȡ��֬��ֽһ��, ����ԲͲ״ ( ��ֱ����С�ڳ���Ͳ�ھ�), �ײ���������(��Ҫʱ����������), װ����Ʒ, �Ͽڸ���֬��, �Ա�֤����Һ���ȵؽ�����ȡ�
˼����4����Ҫ��Ϊ�������ܼ��Ľ�������������ȡЧ�ʡ�
˼����5����Ҫ��Ϊ��ʹ�ܼ����ȵĽ����Ҷ��
˼����6�𣺷�����ʯ�ҿ����кͲ�Ҷ�еĵ����ᣬ�����������ˮ�֡�
˼����7������������ˮ�֣�������ʼʱ��������һЩ��������Ⱦ����Ͳ�Ʒ��
˼����8�����������ϸ��Ǵ���С����ֽ��Ϊ�˱����������Ŀ�����������������У�ֽ�ϵ�С��Ӧ��֤����ͨ����©����������Ϊ��ֹ�����������ݳ���
˼����9���¶�̫�ߣ������±��������ֽ̿����һЩ��ɫ����Ҳ�ᱻ��������Ӱ���Ʒ������������������ʱ��������Ӧ�ϸ���ơ�
˼����10�𣺿�������������������ɱ�������Լ��������ɰ�ɫ������
˼����11�𣺺��ɫ�ij�����
˼����12�𣺿�����ɱ��������⡢����ص������������������ļ�ż���ʣ�������ˮԡ���ɣ���õ��ɫ���������백���ü�������ɫ������李��÷�Ӧ�����������������Է�Ӧ��
ʵ��ʮ�� ����ͪ���Ʊ�
һ��ʵ��Ŀ��
1�����ս�������ʹ�÷�����
2����һ����������Һ��ĸ����ȡ�Ȳ�����
3��ͨ���Ʊ�����ͪ ��ѧϰ��~�˷�Ӧ��
����ʵ��ԭ��
����ʵ�鲽����װ��
1�� װ��
2�� ʵ�鲽�� �����Ҫ�ܽ�ʵ�鲽�裬��д����Ӧ��ʵ������
��װ��10 mL��ѹ��Һ©������е����װ�úͻ���������(�϶�ͨ��һ�Ȼ��Ƹ�������Ȼ�����������װ������)��100mL������ƿ��Ѹ�ټ���13 g(0.097 moL)��ĩ״��ˮ���Ȼ�����16 mL(Լ14 g��0.18mol)��ˮ�����ڽ����½�4 mL(Լ4.3g��0.04mol)�����Ե�Һ©�������μӵ�������ƿ��(�ȵ��뼸�Σ�����Ӧ�������ټ����μ�)�����������ĵμ��ٶ���ʹ������ƿ����Ϊ�ˡ������(Լ��10min)������Ӧ�Ժͻ����ڷ�ˮԡ�н��������ֱ���������Ȼ��������ݳ�Ϊֹ��
����Ӧ�������ȴ�����£��ڽ����µ���18 mLŨ�����35g������ձ���(��ͨ�ڳ��н���)�������й��岻����ɲ�������Ũ����ʹ֮��ȫ�ܽ⡣�������ת���Һ©���У��ֳ��л���(��һ�㣿)��ˮ���ñ���ȡ2��(ÿ��8mL)���ϲ��л��㣬������15 mL10%�������ơ�15 mLˮϴ�ӣ�����ˮ����þ���
����ˮԡ��������ձ���Ȼ����ʯ�����ϼ�����ȥ�����ı������¶�����140������ʱ��ֹͣ���ȡ��������ÿ���������(Ϊʲô��)�����ռ�195~202����֣����أ�����ԼΪ4.1 g������85%)��������ͪΪ��ɫ����״Һ�壬bpΪ202�棬
�ġ�ע������
1����Ӧװ��Ҫ�������AlCl3��ˮ�����ֱ���������ˮ�⣬�����á�
2��AlCl3 Ҫ���飬�ٶ�Ҫ�졣
3������ʦ�����ܿ�������װ�á�
4������ϡHClʱ����ʼ���Σ��죻ϡHCl��1:1�����䣩����ԼΪ140mL��
5������װ�ã�Լ20������������Һ�����䣬200mL���ر�ע���ֹ������
�塢ʵ����
1��������ʣ����۲����Դ�����Ϊ���㣩
2��ʵ������У���ɫ����α仯�ģ����û�ѧ����ʽ��ʾ��
��������������
1����ʵ��ΪʲôҪ�ù����ı���AlCl3 ��
2����Ӧ��ϣ�����HCl�������ɣ������������δ���꣬ԭ����ڣ���ʵ�������к�Ӱ�죿
ʵ��ʮ�� ���ȵ��Ʊ�
һ��ʵ��Ŀ��
1��ͨ�����ȵ��Ʊ�ѧϰ�ص�����Ӧ��ż�Ϸ�ӳ��ʵ�������
2�������������ؽᾧ��ԭ���Ͳ�����
����ʵ��ԭ��
����������ҩƷ
���������ᡢ5%NaOH��NaNO2��Ũ���ᡢ�����ᡢN��N-����������10%NaOH��NaCl���Ҵ������ѡ�������������ѭ��ˮ��ձ�
�ġ�ʵ�鲽��
1�������������ص��ε��Ʊ�
�� ��100mL�ձ��У�����2g���������ᾧ�壬��10mL5%NaOH����ˮԡ�����ܽ⡣
�� �������£���0.8g NaNO2���ܽ���ڽ����£������ܽ��������װ��13mL�����ˮ��
2.5mLŨ�����С�
�� ʹ�¶ȱ�����50C���£�����Ӧ���������ڱ�ԡ����15min��
2��ż��
�� ��һ֧�Թ��м���1.3mL N,N-����������1mL�����ᣬ��ϡ�
�� �ڽ����£�����Һ�����ӵ�������ȴ�ص����У�����10min��
�� ��ȴ���裬��������15mL10%NaOH��Ϊ��ɫ��
�� ����Ӧ����������ڣ��ܽ�����䣬���ڱ�ˮԡ����ȴ��ʹ����ȫ�����½ᾧ���������ռ��ᾧ��
�� �ñ���NaCl��ϴ�ձ����Σ�ÿ��10mL�����ô˳�ϴҺϴ�Ӳ�Ʒ��
3������
�� ����ƿ��ͬ��ֽ�Ƶ�װ��75mL��ˮ���Ƚ��裬ȫ�ܺ���ȴ�����£���ԡ��ȴ�����Ƚᾧȫ�����������ˡ�
�� �����������Ҵ�������ϴ�Ӳ�Ʒ��
�� ��Ʒ�������(����Լ2.5g������75%)��������ʡ�
4������
�ܽ�������Ʒ���Ӽ���ϡHCl��Ȼ����ϡNaOH�кͣ��۲���ɫ�仯��
�塢ʵ��ע������
1������������Ϊ���Ի��������ǿ�ڼ��ԣ�����������ó��ζ������������ó��Ρ�
2���ص��������У�Ӧ�ϸ�����¶ȣ���Ӧ�¶�������5�棬���ɵ��ذ�����ˮ��Ϊ�ӣ����Ͳ��ʡ�
3������ֽ����ɫ���貹������������Һ��
4���ؽᾧ����ҪѸ�٣��������ڲ���ʼ��ԣ����¶ȸ�ʱ�ױ��ʣ���ɫ������Ҵ�������ϴ�ӵ�Ŀ����ʹ��Ѹ�ٸ��
������������
1���ڱ�ʵ���У��ص��ε�ֲ��ΪʲôҪ������0~5���н��У�ż�Ϸ�ӦΪʲô�������Խ����н��У�
2����ֲ���ص����м����Ȼ���ͭ������ʲô���Ľ����
3��N,N-�����������ص���ż��Ϊʲô�����ڰ����Ķ�λ�Ϸ�����
ʵ���ʮ ����ˮ������Ʊ�
һ��ʵ��Ŀ��
1������ˮ������������Ӧ��ԭ���Ͳ�����
2����Ϥ�����л����ؽᾧ�ᴿ������
����ʵ��ԭ��
����ˮ������Ʒ��Ϊ��˹ƥ�֣�Ϊ���õ�������ʹҩ���Ʊ�����ˮ������õķ����ǽ�ˮ���������������ã�ͨ����������Ӧ��ʹˮ��������з��ǻ��ϵ���ԭ�ӱ�������ȡ������������ˮ���ᡣΪ�˼��ٷ�Ӧ�Ľ��У�ͨ����������Ũ������������Ũ������������ƻ�ˮ����������Ȼ�����ǻ����γɵ�������Ӷ�ʹ�������ý�����ɡ��䷴Ӧʽ���£�
�����õ����Ǵ�������ˮ���ᣬ���з�Ӧ���������δ���õ�ԭ�ϡ������ȣ����뾭�������������ܵõ���Ʒ��
������̬�л��ﳣ�õķ�����ѡ����ʵ��ܼ������ؽᾧ���ؽᾧ��һ�����Ϊ��
�ٽ��ֲ�Ʒ�����ȵ��ܼ����Ƴɱ�����Һ���ڳ��ȹ��˳�ȥ���������ʣ��۽���Һ��ȴ��ʹ�ᾧ�ӹ�������Һ��������������������ĸҺ�У��ܳ������ˣ���ĸҺ�н��ᾧ������ϴ�ӽᾧ�Գ�ȥ������ĸҺ�����õĽᾧ�������Ϊ���������ʡ���ʱ�������������ظ���Σ����ܻ�ô�Ʒ��
�ؽᾧ�Ļ���ԭ���ǣ������л������ܼ��е��ܽ�����¶������й�ϵ��һ�����¶������ܽ���������ѹ����ܽ����ȵ��ܼ��дﵽ���ͣ���ȴʱ�������ܽ�Ƚ��ͣ���Һ��ɹ����Ͷ������ᾧ�������ܼ��Ա��ᴿ���ʼ����ʵ��ܽ�Ȳ�ͬ������ʹ���ᴿ���ʴӹ�������Һ����������������������ĸҺ�У�����˳�ȥ�����Ӷ��ﵽ�ᴿĿ�ġ�
�ڽ����ؽᾧʱ��ѡ���ʵ����ܼ��Ƿdz���Ҫ�ģ�����ﲻ��������Ŀ�ġ���Ϊ�ʵ����ܼ�������߱������������ٲ��뱻�ᴿ������ѧ��Ӧ�����ڽϸ��¶�ʱ���ܽ����Ҫ�ᴿ�����ʣ���������ʱֻ���ܽ���������۶����ʵ��ܽ�ȷdz����dz�С��ǰ�߿ɽ���������ĸҺ�в����ᴿ�ᄃ��һͬ������������ʹ�������ȹ���ʱ����ȥ���������ӷ�������ᾧ�����ȥ��
���ѡ��һ�ֺ��ʵ��ܼ�ʱ������ʹ�û���ܼ�������ܼ����ǰѶԴ������ܽ�Ⱥܴ���ܽ�Ⱥ�С�����ܻ��ܵ������ܼ��������ʹ�ã��ɻ������Ľ����
���Ƶ�����ˮ��������Ҵ�һˮ����ܼ������ؽᾧ������ˮ����Ϊ��ɫ��״�ᾧ���۵�Ϊ143�棬����ˮ���������Ҵ����л��ܼ��С�
��������
1������ ��ƿ����¯��ˮԡ�����¶ȼƣ���ƿ�У��ձ�������©��������ƿ����Ͳ��̨�ӣ��������Թܣ�ˮ�ã����ͱã�����ֽ��
2��ҩƷ ˮ���ᣬ��������Ũ���ᣬ95%�Ҵ���1%���Ȼ����� ���顣
�ģ�ʵ�鲽��
1������ˮ������Ʊ� ��100mL�������ƿ�м���6.3g��Լ0.045mol��ˮ�����9mL��Լ0.09mol�����������ټ�10��Ũ���ᣬҡ�ȡ�����ƿ����80 ~ 90���ˮԡ�м��ȣ�ˮ�����ܽ��ά�ַ�Ӧ20min��ʱ����ҡ��ȡ����ȴ�����50mL����ˮ��Ȼ����ƿ���ڱ�ˮ����ȴ15min�����д����ᾧ����ʱ�������ˣ���ֽ������ˮ��ʪ����������������ˮϴ�ӽᾧ���Σ���ɣ����ô��Ƶ�����ˮ���ᡣ��1%���Ȼ�����Һ��������ˮ����Ĵ��ȡ�
2����������ˮ������ᴿ���ؽᾧ �����Ƶ�����ˮ�������50mL�ձ��У�����10mL�Ҵ���մ���ڲ���©������ֽ�ϵľ��嶼ϴ���ձ��У���ˮԡ�ϼ���ʹ���ܽ⡣���ȳ������ˣ���ֽ���Ҵ���ʪ�����Գ�ȥ���������ʡ�����Һ����һ�ɾ��ձ��У�����30mL����ˮ�����������ˮ����ȴ15min�����ᾧƽ���ڱ������ϣ�����100������к��20 min���������ز�������ʡ�
ͬǰ������1%���Ȼ�����Һ��������ˮ����Ĵ��ȡ�
�壮ע������
��Ӧ�¶Ȳ��˹��ߣ��������Ӹ������������ɡ�
����˼����
1���ؽᾧ�ᴿ��ԭ����ʲô��
2��ǰ����������Ȼ�����飬����˵����ʲô��
3�����Ʊ�����ˮ��������У�Ӧע����Щ������ܱ�֤�нϸߵIJ��ʣ�
ʵ���ʮһ ��������Ʊ�
һ��ʵ��Ŀ��
1��ѧϰͨ�������ķ����Ʊ��
2������Ũ�������ˡ��ؽᾧ�Ȳ������ܡ�
����ʵ��ԭ����
����֪���Ʊ��������÷����������������õ�ԭ�Ͽ�����ϩ����RCH2=CH2������(R-CH2OH)��ȩ(R-CHO)�ȣ���������������HNO3��Na2Cr2O7��H2SO4(��K2Cr2O7��H2SO4)��KMnO4��H2O2��CH3COOOH�ȣ�Ҳ�����ô�����������ʵ����HNO3�����������Ʊ�������
����Ӧ ��
�ڷ�Ӧ�У�HNO3��Ϊһ���������ı��������ɳ���̬��[O]
��Ҫ����Ӧ������������ȿɵõ��������ḱ���
Ϊ��ֹ��Щ������϶�����ɣ�����Ʒ�Ӧ���˹��ڼ��ң�����ʹ�õ���������Ũ�Ȳ��˹��ߣ�����ͨ����ʹ���������������⣬��Ӧ�¶�Ҳ�����½��У��¶Ⱥ�Ũ�ȹ��߽��������
������Ӧ�IJ������Ρ����Ρ������Ρ������Ρ��Լ��������ζ�����Ϊ��HNO3��������ʱ�Ĵ������Լ�ǿHNO3���������á�
�ڽ����ʵ�鵱�У����ʹ�����ϵ�������������ӦӦ����50��60���½��в��ô���ʱ����ӦӦ����80��100��֮����С�
���������Լ�����������������
����
������
��״
d
����
m.p/��
b.p/��
�ܽ��
ˮ
�Ҵ�
����
������
90
��ɫ����
0.9624
25.2
161
����
����
����
������
198
��ɫ�ᾧ��ĩ
1.366
196
152
330.5
����
����
����
����
63
��ɫҺ��
1.42
~42
86
��
��
��
�ġ�ʵ�鲽��
1����125mL��������ƿ��װ�õ�Һ©�����¶ȼƺͻ��������ܡ����������Ͻ�һ��������װ�ã���10%������������Һ��Ӧ�����в����Ķ����������塣
2����������ƿ�м���6mL50�� ������(0.06mol)�����������(Լ0.01g)��
3����ˮԡԤ��������Һ��50�棬���ң���֧ˮԡ���Եο��ȵ���5~6�λ��Ѵ�����ͬʱ����ҡ��������Ӧ��ʼ�ų������������壬Ȼ�������μ����ಿ�ֵĻ��Ѵ�������Ϊ2 mL(Լ0.02mol)�����ڵμ��ٶȣ�ʹƿ���¶�ά����50~60��֮�䣨�ڵμ�ʱ��������ҡ�������μ����Լ��5min��
4���μӽ�����������80~90���ˮԡ���ȣ���Ӧ10 min������������ɫ����ų�Ϊֹ��
5����ȴ��Ӧƿ��ʹ����������
6�����ˣ���3mL��ˮϴ�ӹ��壬ѹ��ˮ�ݡ�
7������, ���أ�����Լ1.7g������58%��, ������ʡ�
��������Ϊ��ɫ��״���壬 m.p. 153�档
�塢������Ӧע������⣺
1������ȡ������ʱ����ʹ�������������Ͳ����Ϊ���ἤ�ҷ�Ӧ�����������⡣��
2���������ڽϵ��¶���Ϊ��״���壬�ۻ�ʱΪճ��Һ�壬�������������ȡ���������ˮ��ϴ��Ͳ��һ�������Һ©���У������ȿɼ�������ճ����ʧ��Ҳ������ˮ�Ĵ��ڶ����ͻ��������۵㣬�����ڵμӹ����нᾧ������Һ©����
3������Ӧǿ�ҷ��ȣ��������в���һ�μ�����࣬����Ӧ̫���ң���������ը��
����˼����
1���Ʊ��Ѷ���ʱ��Ϊʲô�����ϸ���Ƶμӻ��Ѵ����ٶȺͷ�Ӧ���¶ȣ�
�𣺸÷�ӦΪǿ���ȷ�Ӧ�������Ѵ��ĵμ��ٶ�̫�죬��Ӧ�¶�����̫�ߣ���ʹ��Ӧʧ�أ������Ѵ��ĵμ��ٶȹ�������Ӧ�¶�̫�ͣ���Ӧ�ٶ�̫������ʹδ���õĻ��Ѵ�����������
2����ʵ��Ϊʲô������ͨ����н��У�
�𣺶�������Ϊ�ж����°����ʣ�Ӧ������ɢ���ڡ�װ��Ӧ���ܣ������ͨ����н���ʵ�顣�緢���������ݳ���Ӧ������ͣʵ�飬���������ܺ����¿�ʼ��Ӧ��
ʵ���ʮ�� ��������Ʊ�
һ��ʵ��Ŀ��
1��������ϵʵ�����շ�����ͨ�������Ʊ������ԭ����ʵ���Ҳ��������̼ױ������Ʊ��������ԭ����������
2�����ռ��Ȼ��������ˡ����ˡ��ؽᾧ�Ȳ������ܣ������ؽᾧ���ᴿ������̲ⶨ�۵��жϴ��ȡ�
����ʵ��ԭ��
����ʵ��������ҩƷ
���������ܡ�Բ����ƿ��ѭ��ˮ��ձá���������������ԡ���ȣ��ױ���KMnO4��Ũ���ᡢNaHSO3���չ�����ֽ�ȡ�
�ġ�ʵ�鲽��
1����250mLԲ����ƿ�м���100mLˮ��2.7mL�ױ�(0.025mol)�����������ӣ����뵽��ԣԡ���
2��װ�ϻ��������ܣ��������������������������ڡ�
3�����������Ͽڷ�������8.5��KMnO4(0.054mol)����������1.5~2h������Һ������������Ϊֹ��
4�����ȳ��ˡ�������Һ����ɫ�ɼ���NaHSO3��������ɫΪֹ����
5����ȴ�����¡�����Һ�μ�ŨHCl��������Ϊֹ���øչ�����ֽ���飩��
6�����ˡ�������أ�Լ1.7g������������ʡ�
7����ˮ�ؽᾧ��
8���������۵㡣
�塢˼����
1����������Ӧ�У�Ӱ�챽�����������Ҫ��������Щ��
2��������Ӧʱ����Ӧ�м�NaHSO3��Ŀ����ʲô��
3���ױ��ø�����������ܷ��Ƶñ���ȩ��Ϊʲô��
ʵ���ʮ�� �������Ʊ�
һ��ʵ��Ŀ��
1�� ������������ԭΪ������ԭ����ʵ���ҷ���
2������ˮ��������ͼ�����Ļ�����������Ϥ��ȡ���뼼�ܡ�
����ʵ��ԭ��
��������ȡ���������κ�ֱ�ӵķ�������������NH2�����뱽���ϡ����Ǿ�����ӵķ�������ȡ���������������ﻹԭ���Ʊ���������Ҫ������ʵ���ҳ��õķ���������������Һ���ý������л�ѧ��ԭ������������������ԭ�����������Ҳ���������������������ᷨ��
����ʵ��������ҩƷ
������ƿ�����������ܡ� ��ѹ��Һ©������е��������Y�ܣ��¶ȼƣ���Һװ©����ˮ��������װ�ã���ԡ���ȣ�����������ԭ���ۡ������ᡢ���ѡ��������ơ����εȡ�
�ġ�ʵ�鲽��
a�� ��һ��100mL��Բ����ƿ�У�����9g������4mL��������װ�ϻ���װ�ã���ȡ20mLŨ���ᣬ�����δ������ܿڼ�����ƿ�ڲ�����ҡ����ӦӦ��������Ӧ̫���ң�ƿ�ڻ�������ʱ������ƿ������ˮ��Ƭ�̣�ʹ��Ӧ������
b�������е�����������ƿ���ڷ��ڵ���ˮԡ�м���30min��ʹ��ԭ������ȫ��Ȼ��ʹ��Ӧ����ȴ�����£���ҡ������������50%NaOH��Һʹ��Ӧ��ʼ��ԣ�
c�� ����Ӧƿ��Ϊˮ��������װ�ã�����ˮ��������ֱ����������ҺΪֹ��
d�������Һ�������©���У�������ֱ�����
e��ˮ������Ȼ���3~5gʹ�䱥�ͺ���20mL���ѷ�������ȡ���ϲ��ֱ�����������ȡҺ������״�������Ƹ��
f���������Ļ��ҺС�ĵ���������50mL������ƿ�ڣ�����ˮԡ����ȥ���ѣ�Ȼ����ÿ��������ܣ���ʯ�����ϼ��ȣ��ռ�180~185�����֣����أ�����2.3g������65%����
�塢�����ص㼰ע������
1����ʵ����һ�����ȷ�Ӧ����ÿ�εμ�������ʱ����һ�����ҵķ�Ӧ��������Ҫ����������
��ֽ��衣
2��������Ϊ��ɫ��״�� ���������Һ�У���ɫ��״����ʧ����ת������ɫ���飬��ʾ��Ӧ����ȫ��
3����Ӧ���Բ����ƿ��ճ���ĺں�ɫ���ʣ���1:1����ˮ��Һ���ȳ�ȥ��
4����20��ʱÿ100gH2O�п��ܽ�3.4g�����Ӵ���Ϊ������
5����ʵ������״NaOH�����ԭ���� CaCl2�뱽���γɵķ��ӻ����
6����Ӧ���ڵ������������ụ�����ܣ���������Һ����������۽Ӵ�������٣���˳����ҡ��Ӧ���ʹ��ԭ����˳�����еIJ����ؼ���
����˼����
1������ʲôԭ����ѡ��ˮ��������ѱ����ķ�Ӧ������з��������
2���������Ƶõı����л����������������ᴿ��
3����Ӧ����ʱ����������Ӧ������ɣ������飬�����뷴ӦҺ����������ҡ������ȫ�ܽ��ʾ��Ӧ����ɣ�Ϊʲô��
ʵ���ʮ�� ���������Ʊ�
һ��ʵ��Ŀ��
1�� ͨ�����������Ʊ�����Է�����ȡ����Ӧ�����⡣
2������Һ������ѹ����ͻ�е�����ʵ�������
����ʵ��ԭ��
������Ӧ���Ʊ������������������Ҫ������Ҳ����Ҫ����ȡ����Ӧ֮һ�������������������У�ͨ����Ũ�����������Ũ�������ã�������ԭ�ӱ�����ȡ����������Ӧ�����������������������ṩǿ���ԵĽ��ʣ����������������ӣ�N+O2�������ɣ�������������
���Լ���������Ӧͨ���ڽϵ͵��¶��½��У��ڽϸߵ��¶����������������������������ԭ�ϵ���ʧ��
����ʵ��������ҩƷ
���������ܡ�����Բ����ƿ����ѹ��Һ©������е��������Y�ܣ��¶ȼƣ���Һװ©������ѹ����װ�ã���ԡ���ȣ�����Ũ���ᡢŨ���ᡢ�������ơ���ˮ�Ȼ��Ƶȡ�
�ġ�ʵ�鲽��
1������ͼװ��װ�á�
2������7.3mL����(0.11mol)��10mLŨ����(0.18mol)��ϳɵĻ�������ѹ��Һ©���У�����Բ����ƿ�з���8.9mL��(0.1mol)��
3���������裬�������������뷴Ӧƿ(��ͨ���������Ͽڽ�����ȴ�Ļ����10~12�μ�����ƿ��)�����Ʒ�Ӧ�¶���50��55�档�������¶ȣ�������ˮԡ��ȴ��ƿ��
4��������Ϻ���ƿ����ˮԡ�м�����60~65�沢����30min����Ъ������ƿ����Ӧ����������£��÷�Һ©��������Ͳ���㣬��������������õ��������ˮ������������Һϴ�ӣ�����ˮϴ�����ԣ��������������������ˮ�Ȼ��Ƹ��
5����50mL������ƿ�����ÿ��������ܣ�ʯ�����ϼ��ȣ����ռ�204~210�����֡�
�塢�����ص㼰ע������
1�����������������Ķ��Խϴ����Դ�������������ʱҪ�ر�С�ģ��粻������Ƥ����Ӧ�����������Ҵ�ϴ�����÷�������ˮϴ�ӡ�
2��ϴ��������ʱ���ر���NaOH���ɹ�������������ʹ��Ʒ�黯���Էֲ㣬����������ɼ������NaOH��NaCl������Һ�μ����ξƾ�����Ƭ�̼��ɷֲ㡣
3�����������ƿ�е��������ڸ���ʱ�������ҷֽ⣬�������Ʒʱ�������ɻ�ʹ�¶ȳ���114�档
4��������Ӧ��һ�����ȷ�Ӧ���¶Ȳ��ɳ���55�档
����˼����
1�� ��ʵ��ΪʲôҪ���Ʒ�Ӧ�¶���50��55��֮���¶ȹ����˸���ʲô���ã�
2�� �ֲ���������ˮ����Һ��ˮϴ�ӵ�Ŀ�ĺ��ڣ�
ʵ���ʮ�� �����ѵ��Ʊ�
һ��ʵ��Ŀ��
1�����մ����Ӽ���ˮ�Ʊ��ѵķ�Ӧԭ����ʵ�鷽����
2��ѧϰʹ�÷�ˮ����ʵ�������
����ʵ��ԭ��
����ҩƷ������
ҩƷ����������Ũ���ᣬ��ˮ�Ȼ��ƣ�5%�������ƣ������Ȼ���
������Բ��������ƿ�����������ܣ���ˮ�����¶ȼƣ���Һ©��������ƿ��
�ġ�ʵ��װ��
�塢ʵ�����
��100mL������ƿ�У�����31mL������(0.34mol)��5mLŨ����ʹ��������ӣ�һ��װ���¶ȼƣ��¶ȼƲ���Һ�����£���һ��װ�Ϸ�ˮ������ˮ�����϶˽�һ���������ܡ����ڷ�ˮ���ڷ���һ������ˮ����һ��������������Ȼ������ƿ������ԡ��������������������������У����з�ˮ����Ӧ�в�����ˮ���������ռ��ڷ�ˮ�����²㣬�ϲ��л��������ˮ��֧��ʱ�����ɷ�����ƿ����Լ��1.5h������ƿ�з�ӦҺ�¶ȿɴ�134~136�档����ˮ��ȫ����ˮ����ʱֹͣ��Ӧ������ӦҺ��ȴ�����º���ʢ��25mLˮ�ķ�Һ©���У������ҡ�����ú���ȥ�²�Һ�塣�ϲ�ֲ���������12mLˮ��8mL 5������������Һ��8mLˮ��8mL
���͵�������Һϴ�ӣ���1g��ˮ�Ȼ��Ƹ�������IJ�������25mL����ƿ�������ռ�140~144��C��֡�
����˼����
1�� ��ε�֪��Ӧ�Ѿ��Ƚ���ȫ��
2�� ��Ӧ����ȴ��ΪʲôҪ����25mLˮ�У�������ϴ��Ŀ�ĺ��ڣ�
ʵ���ʮ�� 2������2���������Ʊ�
һ��ʵ��Ŀ��
1�� �˽�Grignard�Լ����Ʊ���Ӧ�úͽ���Grignard��Ӧ��������
2�� ѧϰ�綯������İ�װ��ʹ�÷�����
3�� ���̻�������ȡ������Ȳ������ܡ�
����ʵ��ԭ��
±�����������þ����ˮ�����з�Ӧ��������±��þ���ֳ�Grignard�Լ�����Grignard�Լ������ʻ�������ȷ����˼ӳɷ�Ӧ����ӳɲ�����ˮ�ֽ�ɵõ�������
����Ӧʽ��
���Լ��� 3.1g��0.13mol��þ����17g��13.5mL��Լ0.13mol�����嶡�飬7.9g��10mL��0.14mol����ͪ����ˮ���ѣ����ƣ������ѣ�10%������Һ��5%̼������Һ����ˮ̼���
����ʵ��װ��
�ġ�ʵ�鲽��
1���������廯þ���Ʊ�
��ʵ��װ��ͼװ������������������������������ƿ��Ͷ��3.1gþ����15mL��ˮ���Ѽ�һС����Ƭ���ں�ѹ��Һ©���л��13.5mL���嶡���15mL��ˮ���ѡ�
����ƿ�ڵ���Լ5mL���Һ�������Ӻ���Һ����״̬�������ɫ��ʧ������������Ӧ��������ˮԡ���ȡ���Ӧ��ʼ�ȽϾ��ң���Ҫʱ������ˮԡ��ȴ������Ӧ���ͺ����������϶˼���25mL��ˮ���ѡ��������裨���ְ��������������ͬʱ����������Ŧ��������ת�٣�����������������嶡�飭��ˮ���ѻ��Һ�����Ƶμ��ٶ�ά�ַ�ӦҺ����״̬��
�μ���Ϻ�����ˮԡ�ϻ���20min��ʹþ������������ȫ��
2��2������2���������Ʊ�
�������ƺõ�Grignard�Լ��ڱ�ˮԡ��ȴ�ͽ����£��Ժ�ѹ��Һ©���е���10mL��ͪ��15mL��ˮ���ѵĻ��Һ�����Ƶμ��ٶȣ���ʹ��Ӧ�������ҡ�������������¼�������15min����Һ�п����а�ɫճ��״������������
����Ӧƿ�ڱ�ˮԡ��ȴ�ͽ����£��Ժ�ѹ��Һ©���з�������100mL10%������Һ���ֽ������ӳɲ����ʼ�����������Ժ���ӿ죩�����ֽ���ȫ����Һ�����Һ©���У��ֳ��Ѳ㡣ˮ��ÿ����25mL������ȡ���Σ��ϲ��Ѳ㣬��30mL5%̼������Һϴ��һ�Σ���Һ������ˮ̼��ظ��
װ������װ�á��������Ĵֲ�������Һ��������С��ƿ�У�����ˮԡ��ȥ���ѣ�����ʯ������ֱ�Ӽ���������Ʒ���ռ�137��141����֡�
�塢ʵ��ؼ�����
1�� �ϸ������װ��ʵ��װ�ã��綯��������봹ֱ��ת��˳����
2�� Grignard�Լ����Ʊ���������������
3�� ��Ӧ��ȫ����Ӧ���ƺõμ��ٶȣ�ʹ��Ӧƽ�Ƚ��С�
4�� ����������������ҽ���������Һ������ȫ��
ʵ���ʮ�� �ڶ�������������Ʊ�
һ��ʵ��Ŀ��
1���˽��ڱ���������������Ʊ�ԭ���ͷ�����
2��ѵ����ѹ�����������ˮװ�õIJ�����Ӧ�á�
����ʵ��ԭ��
�ڱ������������������Ϊ���ܼ�ʹ�ã���Ϊ���ܼ�DBP�������������ᡢճ�����Ⱦ�ϡ�ӡˢ��ī��֯������������ ������ɫ��Һ�壬���з�����ζ�����ӷ�����ˮ�е��ܽ��0.03%��25�棩�Զ�����֬�����к�ǿ���ܽ�������
������������ѵ������������װ�ú���Ҫ���̣�
��ѹ�����������ˮװ�õIJ�����Ӧ��
ʵ������
������
�ڱ���������
��ӦҺ
���
��Ʒ
��Լ3.7g��
H2SO4
��ˮ������
����Լ25min
��Һ
�к͡�ϴ��
����
��ѹ����
���������衿
1����7.5mL��������3g�ڱ�����������4��Ũ�������25mL������ƿ��ҡ�Ⱥ�̶��ڲ���ƽ̨�ϡ�������ƿ��װ���¶ȼƣ���ƿ��Լ0.5cm������ˮ�����������µ�һ����������ס���������Ͽڽ�װ���������ܡ�����ˮ�������м���ˮ��֧�����1.5cm����
2��С�������ƿ���¶Ȼ������������¶�����140��ʱ����Լ��25min����ֹͣ���ȣ���ƿ���¶Ƚ���50������ʱ����ӦҺת���Һ©������10mL 5%̼������Һ�кͷ�ӦҺ���ֳ�ˮ�㡣���ñ���ʳ��ˮϴ��2�Ρ����ֳ�ˮ�㡣�л�����������ˮ�����Ƹ����ת��10mLԲ����ƿ�������ȳ�ȥ���������������ټ�ѹ���� �ò�Ʒ3.7g������Ʒ�е�340�棬 =1.4911 ��
�ġ�ʵ��ؼ���ע������
1����������ˮ���γɹ��л�����ˮ������ˮ���������ϲ�Ϊ���������²�Ϊˮ��Ӧע����ݷ�Ӧ������ˮ�����жϷ�Ӧ���еij̶ȡ�
2����Ӧ�¶Ȳ��ɹ��ߣ��������ɵIJ����������������ֽ�
3���к�ʱӦ���պü�������������Ӱ����﴿�ȼ����ʡ�
�塢��Ҫ�Լ�����Ʒ������������������ֵ��
�� ��
������
�� ״
�� ��
�۵��
��
�ܽ�ȣ���/100mL�ܼ�
ˮ
��
��
�ڱ������������
278.34
��ɫ��Һ��
1.4911
340��
0.03%��25�棩
������Ʒ��״����ۡ�������������������ֵ���գ�
��ɫ��Һ�壬���з�����ζ�����ӷ�
�ߡ����ʼ��㣺
�ˡ����ʸ�Ҫ
1�����㱾��ʵ�鷴Ӧ����Ӧ���ɵ�ˮ�������жϷ�Ӧ���еij̶ȡ�
2����Ӧ���п��ܷ�����Щ����Ӧ��
3�����ֲ����кͳ̶Ȳ������ԣ��Ժ��������ʲô����Ӱ�죿
�š���Ҫ�Լ����������
�ڱ�������������������Ũ����
�����ʵ��
�����Ŀ
�ο�ʵ���ʮ�����ɼױ������Ʊ�������ķ����������2-�ȼױ���3-�ȼױ���4-�ȼױ���2-��ױ���3-��ױ���4-��ױ���2-�����ױ���3-�����ױ���4-�����ױ��Լ���Ӧ��ȡ���ұ������ȼ��鼰���״�����һ��������Ϊ��Ӧ������Ʊ���Ӧ�ı������ȡ�������ᡣ
Ҫ��
�������ϣ���Ʒ�����ʵʩ�Ʊ������������Բ�����Ʊ������롢�ᴿ��������ȫ���̡�
��ʾ
1��ȷ��ʵ���ģ��������С�������������ϳɣ����Ʋ��������
2�� ѡ���������������������������ת�ƴ�������ѡ�������������ø������Ϊ��������Ӧȷ���������ԡ����Ի����ڼ��Խ�����������
3�� ����ȡ������λ�á����ࡢ��������Լ����ϵ�ȡ�����Է�Ӧ�Ƿ����Ӱ�졣
4�� ʲô�������ϱȣ��ܱ�֤��Ӧ����ȫת���ɲ��
5�� ��Ӧ���Ƿ����ִ���ȩ��ͪ�ȸ�����������ٸ������η��룿
6�� ������������������
7�� �������̿�����ϸ����������˳�����ˣ�
8�� ѡ��Ӧװ�ã������Ĺ����Σ�
9�� ѡ���������ķ�����:���۵㡢��ѹҺ��ɫ�ס�TLC������IR�ס���1H-NMR�Ⱦ��ɡ�
10��д��������ʵ�鱨�档
˵��
ָ����ʦ�ɸ���ʵ���ҵ�������ָ��ѧ��ʹ�õ�ԭ�ϣ�������ָ��ѧ������ʵ�飬Ȼ��Աȡ����ۡ��ܽ�ʵ������
�����
Ԭ�ı�����. �л���ѧ. ����:�ߵȽ���������, 2000. 173.
����
1�� �ܽ��Լ���ͬѧ��ʵ������������֮��
2�� �������л�ʵ��ȡ�õ�����֤���л���ѧ�е�ͨ�÷�Ӧʽ��
3�� ��ϱ�ʵ��˵���л���ѧʵ�����л���ѧ���ۼ�Ĺ�ϵ��
�о���ʵ��
�л���ѧʵ�����һ����Ҫ�����Ƿ�Ӧ������ʵ���������ȷ������Ҳ��һ��ӵĹ�����һ����ѧ��Ӧ��ʵ�������ɷ�Ӧ��ṹ���������������¶ȵȲ�������������Ӧ����ֱ��Ӱ�췴Ӧ���ת���ʺͲ���IJ��ʡ����˵ķ�Ӧ��������Ҫͨ���������ϣ��������Ƶķ�Ӧ�����ʵ�������Ȼ��ʵ�����Ƚϡ�ɸѡ�������ȷ��������
�����������Ʊ�ʵ���������о�
����
������Ӧ�ǿ���ƽ�ⷴӦ����Ӧ���ᡢ���Ľṹ�����ϱȡ��������¶ȵȶ�Ӱ��ƽ�⡢��Ӧ�����Լ�ת���ʡ�Ҫ�õ������ʵ������轫��Ӧ��֮һ��������ֳ���Ӧ��ϵ��
����ʵ�����
���ĸ�ʵ�飬ǰ����ʵ���ڻ�����Ӧװ������ɣ����һ��ʵ���ڻ�����ˮ��Ӧװ������ɡ�
ʵ��1 ����0.1mol���촼0.1mol��Ũ����5�Σ�
ʵ��2 ����0.1mol���촼0.1mol������Ũ���
ʵ��3 ����0.12mol���촼0.1mol��Ũ����5�Σ�
ʵ��4 ����0.1mol���촼0.1mol��Ũ����5�Σ���5mL��
��ʵ��1��2��3ʱ������ƿ�зֱ����������Ӧ��ͼ�����ʯ����������ʹ��Ӧ����ֻ�������������30min����ȴ����5mL��ˮϴ�ӷ�Ӧ�������ñ���̼������Һϴ�Ӷ��Σ�ÿ����5mL���������5mLˮϴ��һ�Ρ�����5mL����������ﵹ��50mL��ƿ�У������ռ�144����ǰ����֣�����Һ���档���Һ�����������ռ�144����ǰ����֣��ϲ���������IJ���Һ���������ռ�144~150�������������
ʵ��4�ڻ�����ˮ��Ӧװ���н���������û�����ɵ�ˮ�����ˮ���У�Լ30min�����ڷ�Ӧ�����м�ʱ�÷�ˮ���е��л������ط�Ӧ���С���¼��Ӧ�ֳ���ˮ������ǰ����ʵ��ķ�����������5mL�������봿������
�������
1�� �Ƚ��ĸ�ʵ�飬ȷ�����������Ʊ������ŵ�ʵ�鷽����
2�� �����������Ӧ�Ľ���ƽ�ⳣ����
3�� �о�ʵ���м���5mL���Ƿ��Ҫ��
4�� �о����ᶡ�������������������������ĺϳ��������ܽ�����Ʊ�ʵ���һ�����������������
д���о�������ܽᱨ�档
�����
1�� �ܿ��ܣ���ռ������. �л���ѧʵ���ѧָ��. ����: �ߵȽ���������. 1997.80.
2�� ��ϼ���. �л���ѧʵ��. ������ѧԺ�л���ѧ��������. ����: �������������, 1980. 102
�л������������ʵ��
ʵ��Ŀ��
1. �����±����������ȩ��ͪ���ǡ�����������ȸ������Ļ�ѧ���ʵ���ʶ��
2. ���ռ����������ķ�����
ʵ��ԭ��
1. ±�����Ļ�ѧ����
��ȡ����Ӧ��±��������Ҫ��ѧ���ʡ�
��±��������ȡ����Ӧ�У����ڵ������ɺͽṹ��ͬ����Ӧ�����IJ�������Լ���ǿ�������ص�Ӱ�죬��ʹ�䷴Ӧ�����е�������ȡ����Ӧ��SN1����˫������ȡ����Ӧ��SN2��֮�֡�һ����˵���ܼ�ЧӦǿ���Լ�������CN����I����OH���������Լ��������ڰ�˫������ȡ����Ӧ�����̽��С�����������£�ijһ��Ӧ�У������ֲ�ͬ�ķ�Ӧ�����ֿ�����ͬʱ���������ߴ��ھ���״̬����ˣ�������±�����Ļ�ѧ����ʱ��ֻ����ϸ�ط�����Ӧ�����Լ�������Լ��Ľṹ������Ū���ijһ��Ӧ�����ǰ����������̽��еĺ�ijһ±��������һ��Ӧ�еĻ�ѧ������Ρ�
��Ӧ���̲�ͬ������±�����Ļ�ѧ����Ҳ�в�ͬ��
�ڵ�������ȡ����Ӧ�У�����±�����Ļ�ѧ���Դ����ǣ�
��±����>��±����>��±����
��˫������ȡ����Ӧ�У�����±�����Ļ�ѧ���Դ������ǣ�
��±����>��±����>��±����
��������ֻ�ѧ���Դ���
RCl < RBr < RI
��±���������������Ҵ���ҺѸ�ٷ�ӦΪ����ϩ��ʽ±����CH2=CHCH2X����ʽ±���� �о����������������������Ҵ���ҺѸ�ٷ�Ӧ����±��������:
������
��±�������������ķ�ӦҲ�ܿ죻������±�������ڼ���ʱ�������ɳ�������ϩʽ±����CH2=CHX����ʽ±���� ��ʹ�ڼ���ʱҲ��������Ӧ��������±����Ҳ���ѷ�����������ȡ����Ӧ��
2. ���Ļ�ѧ����
��1����������Ʒ�Ӧ���ɴ��ƺ�����
�ǻ��л����⣬���������÷ų�������
��2���������ٴ���ʹ���������Һ��ɫ
�������ٴ��ܱ��ظ���ء�������ػ���ᣨCrO3�������ᣩ���������������崼��ͬ�������²���������
��3����¬��˹��Lucas���Լ���Ӧ�����١��崼
��¬��˹��Lucas���Լ���ZnCl2��Ũ������Һ����Ӧʱ�����ڷ�Ӧ��Ũ��ͼ��Խ����У���Ҫ��SN1���̽��У��崼������Ӧ���ٴ���Ӧ����������������Ӧ������6��̼���µ�ˮ���ԡ�Ԫ����˵���������ɵ��ȴ��鲻����¬��˹��Lucas���Լ�������״����������˳�����6��̼���²����١��崼�ļ���
�ޱ仯
��4��̼����10�����µĴ�����������Լ�������������ɫ���ɫ�����ɽ��������10��̼���µĴ���
����ɫ���ɫ
��5����λ���ǻ�����ijЩ���۽��������������������εĻ��������Cu(OH)2��������ɫ����
����ͭ������ɫ��
��Ũ���������£�������ܱ��ֽ��ԭ���Ĵ���ͭ�Ρ�
3. �ʻ�������Ļ�ѧ����
��1��ȩ��ͪ���ʻ��Լ���Ӧ
ȩ��ͪͬ���ʻ�������������ʻ��Լ�2,4-���������µȷ����˷�Ӧ�����磺
��ɫ
��2��ȩ�ܱ�һЩ���������������Լ���������Һ�������Լ�������
ȩ���������ᣬ���������ף�Tollens���Լ���������������Һ��Ӧ����������������Schiff���Լ���ϳ��Ϻ�ɫ�Ļ����
����Schiff���Լ�
�Ϻ�ɫ
��ȩ������Schiff���Լ���ϳ��Ϻ�ɫ�Ļ������м�������ʱ�������Ϻ�ɫ�Ļ�������ֽ�, �Ӷ���ɫ��ֻ�м�ȩ������Schiff���Լ���ϳ��Ϻ�ɫ�Ļ������������ʱ����ɫ��
ͪ��������෴Ӧ��
ȩ��Ľ�һ�������ͨ����֣�Fehling����Ӧ��Fehling�Լ�������ɫ������֬��ȩ����ʱ����Һ��ɫ���η��������̡��ơ�ש��ɫ�����ı仯����ȩ�����ܽ�һ����������ͭ��ԭΪ����ɫ�Ľ���ͭ������ȩ��Fehling�Լ��˷�Ӧ����˿���֬��ȩ����
��3�������������Ƶļӳ�:
�����ȩ��֬�����ͪ���˸�̼���µ�֬��ͪ�������������ƣ�NaHSO3��������Һ(40%)�����ӳɷ�Ӧ�����ɲ�����40% NaHSO3�Ħ�-�ǻ������ư�ɫ�ᾧ���˾�������ˮ���������л��ܼ������Ҵ˷�ӦΪ���淴Ӧ�����ɵĦ�-�ǻ���������ϡ���ϡNa2CO3��Һ����ʱ����ֽ�Ϊԭ����ȩ��ͪ����ˣ���һ��Ӧ����������ʹ���ȩ��֬�����ͪ��̼ԭ��������8��֬��ͪ��
��-�ǻ�������
��4����·�Ӧ
�ʻ����������һ��Ҫ��Ӧ�Ǧ�-̼ԭ���ϻ�����ķ�Ӧ����-̼��ĦҼ���̼����м�������-�й����ˣ�ȩ��ͪ��-�����һ���Ļ��ԡ��ܽ��Ц�-±����±�·�Ӧ���Ծ��� �ṹ���ʻ���������õ�ļ�����Һ��֮��Ӧ����·�Ӧ�������ɾ���������ζ�Ļ�ɫ��½ᾧ���м��������ڵ�ļ�Һͬʱ��������������ʹ����������Ӧ��ȩ��ͪ����ˣ����нṹ �Ĵ�Ҳ�ܽ��е�·�Ӧ��
���
��5����ȩ����
��ȩ�����Ǧ�-���������һ����Ҫ��Ӧ�����Ц�-���ȩ��ͪ��ϡ��������£�ϡ�����-��ԭ�ӽ�ϣ��γ�һ�����ȶ��ĸ�̼���ӣ������������ӳɵ���һ����ȩ����ͪ�����ʻ���̼ԭ���ϣ�����������ɣ��������������ϻ����Ϸ�Ӧ�����磬����ȩ�ͱ�ͪ�Ľ�����ȩ���Ϸ�Ӧ��
�����ͪ
�������ͪ����ɫ�ᾧ��
m.p.110��112��
4. ���Ļ�ѧ����
��1�����ļ���
���ɿ����ǰ������е�һ������ԭ�ӱ�����ȡ�����õ������������һ������ˮ��Һ�ʼ��ԣ��������������Ρ�
���ļ���ǿ����͵���������������ЧӦ���ռ�λ��������ء����ְ���pKbֵ����4-1��
��4-1 ���ְ���pKbֵ
��
NH3
CH3NH2
(CH3)2NH
(CH3)3N
pKb
(25��)
4.7
3.4
3.3
4.2
9.4
9.2
8.9
��2����˹����Hinsberg����Ӧ
���в����١���֮�֡��������ٰ����尷��������Ӧ�У����ֳ���ͬ���ص㡣��˹����Hinsberg����Ӧ������������һ�������������벮�����ٰ����尷�ġ�
��3����������ķ�Ӧ
����������ķ�ӦҲ�������ȡ��������������IJ�ͬ����ͬ��֬�������������ᣬ��ʹ��0����������Ӧ�ų��������ٰ�������������������尷����Ӧ�����㲮�����������ڵ��£�<5�棩�·�Ӧ���ŵ����������ص��Σ��ص������-�������ã����ɳȺ�ɫ����Ⱦ�ϡ������ٰ����尷������ͬ����������Ӧ��
��Һ �Ⱥ�ɫ����
�ޱ仯
��ɫ��״��
��ɫ���� ��ɫ����
��˿ɼ����㲮�����ٰ����尷��
5. �ǵĻ�ѧ����
������ﰴ��ṹ��ָ���ǻ�ȩ��ͪ�Լ����ǵ��������ijЩ�����ͨ����Ϊ���ǡ�˫�ǺͶ��ǡ�
��1��Molisch��Ӧ����-��������
���������Ũ�������������������ܲ�����ɫ��Ӧ�����磬�����-������Ũ���������¿�������ɫ����Molisch��Ӧ����������Ϊ���������Ũ�����������ɿ�ȩ����������ȣ���һ�����-��������������ɫ�������ɫ������
���ǡ�˫�ǡ�����һ�㶼�����˷�Ӧ���������Dz������˷�Ӧ����ͪ�����ᡢ���ᡢ���ᡢ������ȩ�ᡢ����ȩ�����������ȩ�Ⱦ��������Ƶ���ɫ��Ӧ����ˣ������˷�Ӧ˵���������Ǵ��ڣ��������һ������������ܿ϶������������˷�Ӧ���ȷ֤��������
��2�����Ǽ����а���ȩ�ǻ���˫�ǵĻ�ԭ��
���Ǽ����а���ȩ�ǻ���˫����ˮ��Һ���ܷ��������칹�������ṹ�뻷״�ṹ����һ����ƽ�⡣��ˣ����л�ԭ�ԣ�Ҳ��Ϊ��ԭ�ǣ��ܻ�ԭFehling�Լ���Benedict�Լ���Tollen�Լ�������������
��3�����۷�Ӧ
����������ı��³��ۣ����ɵ��۾���һ�����۵�;��Σ��������۵ľ��κ��۵�ɼ���ijЩ�ǡ�
���۷�Ӧֻ��C1��C2ԭ���Ϸ�����ֻҪC1��C2�����̼ԭ�ӹ�����ͬ���ǣ��������γ���ͬ�����ۡ���ͬ�����ۻ�ѧ�ṹ��ͬ�����Ρ��۵���ܽ��Ҳ������ͬ��
��ͬ���Ǿ��ܿ����γ���ͬ�����ۣ������ķ�Ӧ�ٶ�ȴ��ͬ���������۵�ʱ��Ҳ����ͬ����ˣ������۷�Ӧ��������ͬ���ǡ�
��ѿ������Һ��ȴ���������������Dz��ܳ��ۣ�����ʱ����ȣ����ǻᱻ�Լ��е���ˮ�⣬
���������Ǻ��Ƕ����ۡ��������۵ĵľ��μ�ͼ4-1:
�������� ��ѿ���� ������
ͼ4-1 ���۵ľ���
�ڱ�ʵ�������£��������۵���ɫ���۵㡢�ֽ��¶ȡ���������ʱ��ͱ���������4-2��ʾ��
��4-2 �����ǵ���������һ����
��
������������ʱ�䣨min��
���۽ᾧ��ɫ
�����۵��ֽ��¶�(0C)
������Һ�ı������
����
5
���ɫ
204
-92
������
10
���ɫ
204
+52.5
��ѿ��
�������
��ɫ
+129.0
����
30��ת�����ɣ�
��ɫ
+66.5
��4�����ǵ�ˮ��
���ǡ����ۡ���ά�صȶ��Ƕ��Ƿǻ�ԭ�ǣ����Dz���ʹFehling��Tollen���Լ���ԭ�������������ø�������¿�ˮ��Ϊ���ǣ�����ˮ��Һ�л�ԭ�ԡ���ά�ص�ˮ��ϵ������ѣ�������ͭ���Լ������������������������ά��������������������ɫ������Ϊ���۵�һ�ּ�����
������������Һ������ˮ�����ɷ������ȵ���С�ĸ��ֺ���������ˮ��Ϊ��ѿ�ǡ������ǡ���ˮ����̿��������Һ��������������ɫ���жϡ�
���� �� �Ϻ��� �� ����� �� ��ɫ���� �� ��ѿ�� �� ������
����ɫ�� ����ɫ�� ������ɫ�� (��ɫ) ����ɫ�� ����ɫ��
����Һɫ��
����ˮ�����Ϊ�����ǣ��Ի�ԭ�ԡ�
6. ������͵����ʵĻ�ѧ����
�������Ԧ�-������Ϊ��������ʰ��ᣨNH2CH2COOH���⣬���ఱ���ᶼ��������̼ԭ�ӣ��������ԡ�
��������а�������NH2�����Ȼ�����COOH�������ʣ������Ի�������еȵ�㡣����������ɵ����ʵĻ���������ijЩ�Լ����ÿɷ�����ͬ����ɫ��Ӧ��ijЩ�����ᣨ���ʣ�����ɫ��Ӧ����4-3��
��4-3 ijЩ�����ᣨ���ʣ�����ɫ��Ӧ
��Ӧ����
�Լ�
��ɫ
���Է�Ӧ��
����ͪ��Ӧ
2,4-������������
��Ӧ
���Ʒ�Ӧ
�����η�Ӧ
Millon��Ӧ
��ڷ�Ӧ ����ͪ
sanger
-
ϵͳ����ʵ�鱨��
ʵ�鱨��һ�������Vensim��VentanaInc�����������ṩ��ǿ���Windows�����µı༭����Ϊ�û��ṩ�Ѻõ�ͼ�ν����ǡ�
-
�Ͼ��ʵ��ѧϵͳ����ʵ�鱨��
����ʵ�鱨��γ���ϵͳ�����ον�ʦרҵѧ��������һ������һ����ȵ�1ѧ���Ͼ��ʵ��ѧ���������ѧԺ1234567891011121��
-
ϵͳ����ʵ�鱨��
����ʵ�鱨��γ����ον�ʦרҵѧ��������һ������һ����ȵ�1ѧ���Ͼ��ʵ��ѧ���������ѧԺ
-
ϵͳ����ʵ�鱨��
����ʵ�鱨��γ���ϵͳ�����ον�ʦרҵѧ��������һһ����һ����ȵ�1ѧ���Ͼ��ʵ��ѧ���������ѧԺ
-
ϵͳ���̷���ʵ�鱨��
ϵͳ���̷���ʵ�鱨����������ӱѧ��110061047�ɼ�ʵ��һ����VENSIM��ϵͳ����ѧ����һʵ��Ŀ��VENSIM��һ����ģ���ߡ�
-
��15����ˮ������ʵ��(��)��ʵ�鱨��
�����������C0791202ѧϰ������°���ѧϰ���IJ��ר����רҵˮ��ˮ�繤��ʵ����������ѹ������һʵ��Ŀ��ͨ������ѹ��ʵ��õ��ԡ�
-
�Ͼ��ʵ��ѧϵͳ����ʵ�鱨��
����ʵ�鱨��γ���ϵͳ�����ον�ʦרҵѧ��������һ������һ����ȵ�1ѧ���Ͼ��ʵ��ѧ���������ѧԺ1234567891011121��
- ��15���ľ����ʵ��(��)��ʵ�鱨�漰��
- ��15������ľ����ʵ��(��)��ʵ�鱨��
- �Ϻ����̼�����ѧʵ�鱨�����