天然药物化学实验指导
天然药物化学实验指导
兰州大学天然药物化学教研室
目 录
天然药物化学实验须知
实验一 薄层色谱(TLC)
实验二 从三棵针中提取、分离小檗碱与小檗胺
实验三 从洋金花中提取分离东莨菪碱和莨菪碱
实验四 大黄中蒽醌类化合物的提取分离
实验五 芦丁的提取与鉴定
实验六 甘草酸的提取
实验七 烈香杜鹃挥发油的含量测定
实验八 丹皮酚的提取、分离和制剂鉴别
实验九 预试验
附录一 常用溶剂的物理性质
附录二 常用溶剂的回收及精制方法
附录三 常用干燥剂性能的说明
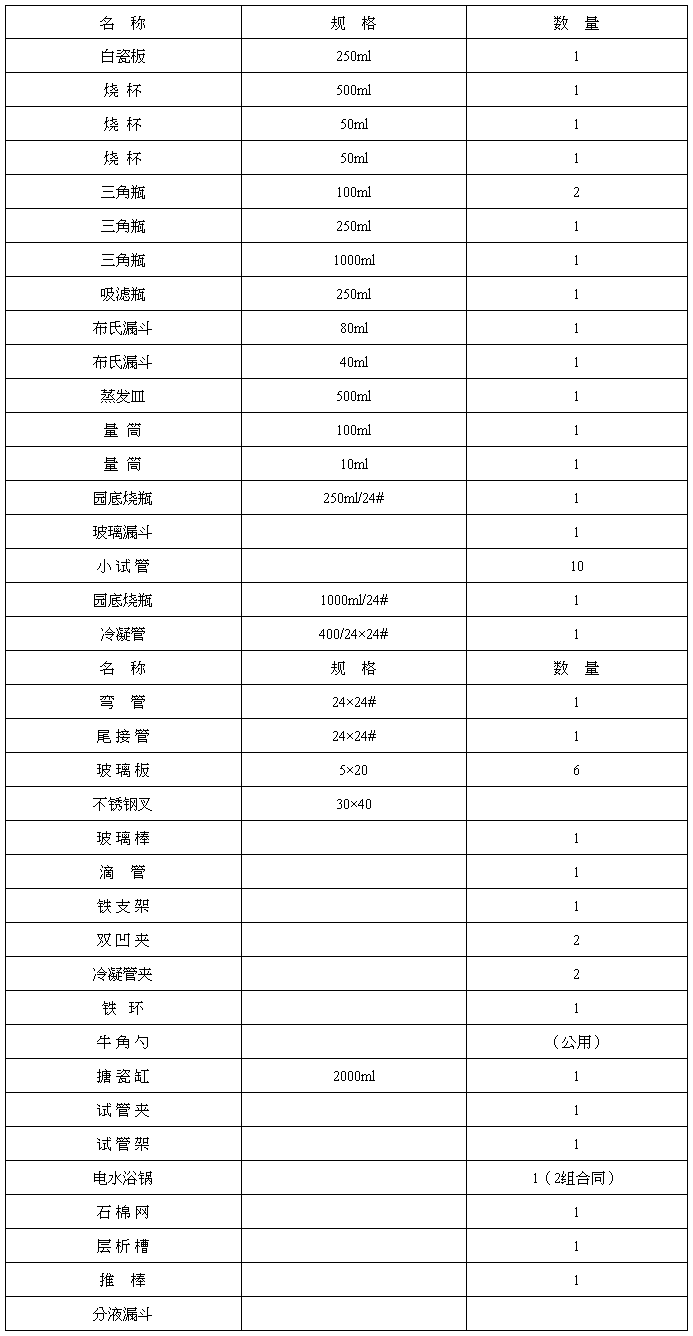
仪器清单
天然药物化学实验须知
一、每次实验以前,必须预习本实验内容,了解其操作程序和理论要点,切勿盲目进行实验,实验时须准备记录薄一本,随时正确记录每一实验应记的项目。如原料用量,实验时发生的现象,反应结果,产品数量、熔点、沸点、产品纯度等,以作写正式报告的依据。
二、实验所得产品,如不供下一实验用,应交给指导老师,并注明品名、数量、组别、姓名、日期。
三、实验规则
1.实验时不能阅读与本实验无关的书籍,不要高声谈笑,也不能擅自离开,严禁在实验室内吸烟及吃东西。
2.实验室要经常保持整齐清洁,桌上不要乱放无用的仪器与药品。火柴杆、废纸等不要乱抛在地上、废酸、碱液应倒入污水缸中,易燃烧及易挥发的废液应倒入水槽中,并立即用水冲掉。
3.药品、仪器都是国家财产,须节约爱护使用,若有损坏,应填写仪器损坏单,报告指导教师,及时领取,并酌情赔偿。
4.试剂、药品、公用用具,使用后立即放回原处,不可乱放,并应注意不可盖错试剂瓶塞,以免污染试剂。
5.装有易燃性液体的瓶子,不得放在灯火近旁。
6.加热乙醇、乙醚、石油醚、苯等易挥发可燃的液体时,应使用装有冷凝管的烧瓶,在水浴上进行加热,加热前应放入沸石1—2粒,或一端封死的毛细管,防止爆沸冲击,若在加热时未放沸石则应冷却后再加,不可热时追加,添加溶剂时应离开火源,稍冷后再添,并应重新加入沸石。
7.实验过程中,万一遇见溶液着火,应立即用石棉布或其他物品把它盖上,使其隔绝空气而熄灭。
8.实验完毕后,应将用过的仪器洗刷洁净放好,并应指定值班人员将实验室打扫干净,污水缸的废液拿到室外适当地方处理埋掉,离开实验室时,应检查门、窗、水电是否关好。
9.严禁将实验室内的仪器、药品携带室外。
以上须知清严格遵照执行。
实验一 簿层色谱(TLC)
薄层色谱是色谱法中应用最普遍的方法之一,具有分离速度快,效率高等特点。适用于微量样品的分离鉴定,天然药物化学成分的研究中得到了越来越广泛的应用和发展。
一、实验目的
l、学习并掌握薄层板的制备和使用方法。
2、了解薄层色谱的原理及应用范围。
二、实验原理
薄层色谱是把吸附剂(或载体)均匀地铺在一块玻璃板或塑料板上形成薄层,在此薄板上进行色谱分离,按分离机制可分为吸附,分配,离子交换和凝胶过滤色谱等。
薄层色谱多数情况是一种吸附层析,利用吸附剂对化合物吸附能力的不同而达到分离,吸附剂吸附能力的大小与化合物极性大小有关。化合物极性大,被吸附剂吸附得牢,Rf值小,反之,化合物极性小,Rf值大。一个化合物在某种吸附剂上Rf值的大小主要取决于展开剂的极性大小,即展开剂极性大,化合物Rf值大,展开剂极性小,化合物的Rf值小。
薄层板根据在制备过程中是干法制板还是湿法制板分为二种。干法制板为软板,湿法制板为硬板。
三、实验内容
(一)薄层板的制备
 1、干法:取150—200目的色谱用中性氧化铝适量,散布在玻璃板上,用一根推捧匀速地从一端向另一端推进,使吸附剂均匀地在玻璃板上铺成一薄层(如图示),即可使用,薄层的厚度取决于推棒下层的塑料圈的厚度,一般以0.25mm为宜。
1、干法:取150—200目的色谱用中性氧化铝适量,散布在玻璃板上,用一根推捧匀速地从一端向另一端推进,使吸附剂均匀地在玻璃板上铺成一薄层(如图示),即可使用,薄层的厚度取决于推棒下层的塑料圈的厚度,一般以0.25mm为宜。

 干法铺层方法
干法铺层方法
干法铺板方法
2、湿法
(1)硅胶G薄层板的制备:称取硅胶G12克,加蒸馏水34毫升,充分搅拌成糊状后,迅速分倒在四块5×20厘米的玻璃板上,铺匀,水平放置,待室温阴干后110℃活化一小时,备用。
(2)硅胶CMC—Na薄层板的制备:称取CMC—Na(羧甲基纤维素钠)0.1克,加l毫升酒精湿润后,溶于17毫升蒸馏水中。立即用力振摇,搅拌(如不溶可加热)全部溶解后,加人过200目筛的层析用硅胶5克,搅拌成糊状,均匀地铺在两块5×20厘米的玻璃板上,水平放置,室温阴干后,于110℃活化一小时,备用。
(二)薄层色谱
l、制板:取150目—200目的层析用氧化铝按上述干法制成薄层板(5×20厘米)一块。
2、点样:取管口平整的毛细管三支分别吸东莨菪碱,其若碱及二者混合的氯仿溶液,点在上述制好的薄层板上,点的直径一般为2—3毫米,点与点之间的距离一般为1.5厘米左右,样品点在离薄层一端为2厘米左右的起始线上,离板边约有l厘米的距离。
3、展开:点样完毕,待溶剂挥干后,用氯仿:丙酮:无水乙醇(8:2:0.5)上行展开,其方法是将薄层板斜放在盛有展开剂的层析槽内,薄板上端有塑料盖垫起使与液面成15。左右的夹角,点有样品的一端浸入溶剂中,深达0.5厘米左右、切勿使溶剂浸没原点,盖好层析槽盖,当溶剂前沿达板的另一端1厘米左右时,即可取出薄层板,标出溶剂前沿位置。
4、显色:取出的薄层板,立即喷洒改良碘化铋钾试剂,使其显色,计算Rf值。
Rf= 起始线到样品斑点中心距离
起始线到剂剂前沿距离
四、实验说明及注意事项
1、制薄层板用的玻璃应干燥、清洁。
2、干法制板时,推捧用力要均匀,中间不要停顿,否则薄层厚度不均匀。
3、点样时,样品浓度不要太大,点样量也不要太多,否则斑点拖尾。若样品浓度太小,可待第一次点样溶剂挥干后再第二次点样。
4、薄层板放于层析槽时,注意切不可将原点浸入溶剂中。
5、层析后的薄层板,取出立即喷洒显色剂,否则溶液挥干后喷显色剂会吹散吸附剂。但湿法制的硬板就不必如此。
6、湿法制的硬板活化的温度和时间可依需要调整:一般检识水溶性成分或一些极性大的成份,所用的薄层板只在空气中自然干燥,即可使用。
五、参考文献
1、中国医学科学院药物研究所:薄层层析及其在中草药分析中的应用,1978。
2、E.菜德雷,M·菜德雷合著:陶义训、马立人、夏寿宣合译:色谱法。
3、全国高等医药院校试用教材:中草药化学,1980。
4、Stahl E,td,Thin Layer Chromatography 2nd edu,1969。
5、E. Heffmann:Chromatography 3nd edu 1975。
实验二 从三棵针中提取、分离小檗碱与小檗胺
三棵针为小檗属(Berberis)植物,种类繁多,是黄连、黄柏的重要代用品。根中含多种生物碱,有季胺类生物碱,如小檗碱(Berberine)1.2—3%,根皮含3—5.5%,少量巴马亭(Palmatine)、药根碱(Jatrorrhigine)、非洲防己碱(Columbamine)、木兰花碱(Magnoflorine),有叔胺碱,如小檗胺(Berbamine)0.45—3.84%,尖刺碱(Oxyacanthine),异粉防己碱(1sotetrandrine)等。
小檗碱是一种常用的抗菌药,对菌痢、肠炎、上呼吸道感染等疾病有良好的疗效。小檗胺有升白、降压、利胆及镇咳等作用,对预防和治疗白细胞减少疗效较好。
一、实验目的
l、了解小檗碱,小檗胺的结构与性质,掌握提取分离方法。
2、了解小檗碱,小檗胺的鉴识反应及薄层色谱鉴别。
二、实验原理
l、小檗碱(Berberine)又名黄连素
黄色针晶,能缓缓溶于水(1:20)、乙醇(1:100),易溶于热水及热乙醇,难溶于乙醚、石油醚、苯及氯仿。
2、小檗胺(Berbamine)
白色或无色结晶,难溶于水,易溶于乙醇中,可溶于乙醚,氯仿、石油醚。
本实验的提取分离原理是:小檗碱、小檗胺的硫酸盐易溶于水,小檗碱的盐酸盐难溶于水,小檗胺的盐酸盐可溶于水,游离的小檗碱为季胺碱可溶于水,游离的小檗胺是叔胺碱,难溶于水。因此,将植物原料用稀H2SO4溶液浸泡,然后用石灰乳调至pHl2左右,小檗碱游离而溶于水,小檗胺含酚羟基成钙盐也溶于水,粘液质及过量硫酸生成不溶性钙盐而沉淀析出,再加NaCl,并用盐酸调pH8左右时,小檗碱成难溶性的氯化小檗碱而析出,小檗胺游离析出,最后利用氯化小檗碱在热水中溶解度较大的性质与小碱胺分离。
三、实验内容
(一)小檗碱与小檗胺的提取分离
取三棵针粗粉100克,置1000毫升三角瓶内,加700毫升0.2%(V/V)的H2SO4溶液浸泡24小时,纱布过滤,再用400毫升酸水同法提取一次,合并两次滤液于搪瓷缸内,搅拌下加人石灰乳至pHl2,静置30分钟,抽滤,滤液中加入计算量(8%W/V)的NaCl,静置,待沉淀完全后,滴加浓HCl至PH8~8.5,于80℃热水中保温半小时,静置,抽滤,所得沉淀于60℃以下干燥,称重,置100毫升烧杯中,加入30倍量蒸馏水,加热至沸,趁热抽滤,滤液内含小檗碱,沉淀为小檗胺的粗品。
(二)精制:趁热向上述滤液中滴加浓盐酸,调pH至2,静置氯化小檗碱沉淀析出,抽滤,60℃以上干燥,称重,计算得率(不得低于0.2%)
上步所得沉淀,60℃以下干燥,称重,置50毫升烧杯中,加20倍量乙醇加热溶解,加入2%活性炭,再加热煮沸5分钟,热过滤,滤液蒸除乙醇,剩l/4量,冷却,滴加蒸馏水至不再析出沉淀为止,用5%NaOH调pH8—8.5,80℃水浴加热5分钟左右,放置,抽滤,干燥,再以10倍量乙醚溶解,过滤,回收乙醚,得白色小檗胺。
(三)鉴识
l、小檗碱的鉴识
(1)取氯化小檗碱水溶液数滴,加浓HNO3 1滴,观察现象,再滴加浓HNO3观察有何变化?解释现象。
(2)取氯化小檗碱水溶液数滴,加锌粒少许,再加浓H2SO4数滴,观察现象并解释。
(3)取氯化小檗碱水溶液数滴;加5%NaOH 2~3滴,再加丙酮数滴,现察现象并解释。
2、小檗胺的定性
(1)取小檗胺结晶少量,置白瓷板上,加5%HC12滴使溶解再加固体硝酸钠使之饱和,移入毛细管中观察。
(2)取l~2粒KMn04置白瓷板上,加5%HCl数滴使溶解,再加少量小檗
胺结晶,观察颜色变化。
(四)薄层色谱
层析板:硅胶G板,110℃,活化半小时。
展开剂:氯仿—甲醇—氨水(15:4:0.5)
样品:氯化小檗碱乙醇液
氯化小檗碱标准品液
小檗胺乙醇液
小檗胺标准品液
显色:碘蒸汽或改良磺化铋钾
四、实验说明及注意事项:
1、用硫酸浸泡时,硫酸的浓度以0.2%为宜,此时生成的硫酸小檗碱在水中溶解度较大,若加入过量,小檗碱就形成酸式硫酸盐,水中溶解度就降低(1:100),影响小檗碱的提取量。
2、冷浸时间不宜过长,次数也不宜过多,否则浸出的杂质量也相对增加,冷浸一般24小时可浸出92%的成分,所以浸出两次即可。
3、在pH~8.5时小檗胺沉淀较完全,但不易凝聚,故80℃保温,使凝聚而沉降。
4、小檗碱精制时调pH至2,是为了使小檗胺等叔胺型生物碱留在溶液中除去,以便得到较纯的小檗碱,操作时若溶液己冷却析出结晶,就应加热成澄明溶液再用盐酸调pH2。
五、参考文献
l、沈阳药学院:植物化学教程1979年
2、贵阳医学院药学系,贵州药讯,1980年5期2页
3、中药声,1979年278页
4、中草药化学,全国高等医药院校试用教材,1980年7月
5、韩秋生等,云南医药,1981.3(56)
6.韩秋生等,药学通报,1981.3(13)
实验三 从洋金花中提取分离东莨菪碱和莨菪碱
洋金花为茄科植物毛花蔓陀罗(Datum innoxia Miller)的花,主要含生物碱,为中麻药方剂中的主药,临床上治疗气管炎有效。
一、实验目的
l、通过本实验掌握生物碱的一般提取分离方法。
2、掌握氧化铝柱色谱分离莨菪碱和东莨菪碱的实验方法,从而了解柱色谱的一般实验操作技术。
3、了解东莨菪碱和莨菪碱的检识方法。
二、实验原理
洋金花中的主要化学成份
l、东食著碱(Scopolamine)又名:莨菪胺、天仙子碱mp59℃(一水合物)。
2、莨菪碱(Hyoscyamine)又名:天仙子胺mp108.5℃
3、莨菪酸(Tropic acid) mp118℃
4、去甲莨菪碱(Norhyoscyamine) mp142℃
5、曼陀罗碱(Meteloidine) mp141℃
本实验是利用游离生物碱及其盐均能溶于乙醇的性质,用乙醇回流提取总生物碱,再利用东莨菪碱和莨菪碱碱性强弱不同,与碱性氧化铝吸附力强弱不同,用氧化铝柱色谱进行分离。
三、实验内容
(一)提取总碱:取洋金花粗粉50克,置1000毫升园底烧瓶中加95%乙醇300毫升,置水浴上回流提取1小时,倾出提取液,药渣再用95%乙醇200毫升不浴回流提取30分钟,倾出提取液,药渣再用95%乙醇200毫升回流提取30分钟,倾出提取液,三次提取液合并,用玻璃漏斗过滤,滤液回收乙醇至无醇味,浓缩液加1%盐酸l00毫升使溶解,抽滤,滤液用氯仿(40,20,20毫升)萃取脂溶性杂质,酸水液用10%NaOH调至pH9一l0用氯仿萃取(60,50,30,20毫升)四次,氯仿液用无水硫酸钠脱水干燥,回收氯仿得总碱。
(二)莨菪碱与东莨菪碱的分离
这两种生物碱我们用碱性氧化铝干柱色谱进行分离,其操作步骤如下:
1、装柱:将色谱柱垂直固定在铁支架上,柱底填一块棉花,打开活塞,称取100—120目碱性氧化铝40克,经漏斗慢慢装入柱中,一边装填,一边用橡皮塞轻轻敲击色谱柱,使其装填均匀紧密,最后使吸附剂表面形成一水平面。
2、上样:将上述所得总生物碱用少量氯仿溶解,用吸管小心将氯仿溶液加在吸附剂表面上,切勿破坏吸附剂平面,为了防止平面被损坏,可剪一直径同色谱柱内径大小一样的滤纸,小心放在吸附剂平面上,或在其上面加1—2厘米厚的石英砂,把氯仿溶液加到滤纸片上或石英砂上。
3、展开(洗脱):加样完毕,立即小心加入氯仿(也应注意防止破坏吸附剂平面),展开洗脱,当溶剂前沿到达柱底部时,用50毫升锥形瓶收集洗脱液,并控制流速,使每分钟流出液为60一80滴,每瓶收集50毫升,更换锥形瓶,编上序号,并在柱上经常添加氯仿,使溶剂面始终高于吸附剂表面,东莨菪碱先洗脱下来,莨菪碱后洗脱下来。
4、溶剂回收,按序号分别回收溶剂后,再氧化铝薄层检查,以氯仿—丙酮—乙醇(8:2:1)展开,改良碘化铋钾试剂显色。相同部分合并。
(三)检识反应及薄层检查
1、生物碱沉淀反应
分别取东莨菪碱,莨菪碱少量,置白瓷板中蒸发到干,加2滴稀HCl溶解,
分别加以下生物碱沉淀试剂:
(1)碘—碘化钾试液
(2)改良碘化铋钾试液
(3)苦味酸试液
观察结果:
2、Vitali反应:分别取东莨菪碱和莨菪碱少量,置于白瓷板中,加发烟硝酸5滴,于水浴上蒸干,放冷后,加10%KOH乙醇溶液2—3滴,观察颜色变化,并加以解释。
3、薄层色谱
吸附剂:碱性氧化铝150一180目,干法铺板。
展开剂:氯仿—丙酮—乙醇(8:2:1)
样品:东莨菪碱氯仿溶液
莨菪碱氯仿溶液
东莨菪碱标准品
莨菪碱标准品
显色剂:改良碘化铋钾,喷洒
四、实验说明及注意事项
l、装柱还可采用湿法:将洗脱剂加到吸附剂中,搅拌成稀糊状,加到层析柱中,使吸附剂依重力自然下沉到柱底部,打开活塞使洗脱剂流出,但要保证溶剂表面始终高于吸附剂表面。干法装柱操作简便、迅速、不受洗脱剂的限制,但柱效不高。
2、柱色谱和薄层色谱用的氧化铝要注意活度,一般Ⅱ—Ⅲ级即可,活度不够就不能将样品中成分分开。
五、参考文献
1、赵承锻Chin.J Physil vol 9.77.1935
2、林启寿等,药学学报,1卷2期95页,1953
3、岛田克卫:药学杂志,vol 75,609,1955
4、中成药研究:1979,l(1—7)
5、中麻通讯:1977.3(18—23)
实验四 大黄中蒽醌类化合物的提取分离
大黄为蓼科大黄属植物掌叶大黄(Rheum Palmatum L),唐古特大黄(R.tanguticum Maxim.)或药用大黄(R.officinale Baill)的根茎,主要含蒽醌类化合物,有泻下和抗菌作用,为常用中药之一。
一、实验目的
l、掌握梯度pH萃取法和提取分离大黄中各种蒽醌甙元的原理及实验方法。
2、了解蒽醌类化合物的颜色反应及色谱检查方法。
二、实验原理
大黄中目前已知的主要成分:
1、大黄酸(Rhein)
mp321℃
2、大黄素(Emodin)mp254~6℃
3、芦荟大黄素(Aloe-emodin)mp223~5℃
4、大黄酚(Chrysophanol)mp193~6℃
5、大黄素甲醚(Physcion)mp203~7℃
其甙有大黄酸葡萄糖甙(Rhein monoglucoside)mp 266℃;大黄素葡萄糖甙
(Fmo din monoglucoside)mp 189℃;芦荟大黄素葡萄糖(Aloe-emod-inmonog-
lucoside)mp 239℃;大黄酚葡萄糖甙(chrysophyanol monoglucoside)mp245℃;大黄素甲醚葡萄糖甙(Physcion monoglucoside)mp 235℃;番泻甙A、B、C、D、E(Sennoside A.B.C.D.E)等。
本实验是根据大黄中的蒽醌类甙元成分能溶于氯仿的性质,采用氯仿提取,又利用羟基蒽醌类化合物酸性强弱不同,用pH梯度法进行分离。具有羧基或多个β位酚羟基的蒽醌可溶于5%碳酸氢钠溶液;具有一个β位酚羟基的蒽醌可溶于5%碳酸钠溶液,只具有α位酚羟基的蒽醌,酸性弱,只溶于氢氧化钠溶液。
三、实验内容
(一)大黄中蒽醌甙元的提取分离
1、大黄中蒽醌甙元的提取:取大黄粗粉50克置于1000毫升园底烧瓶中,加20%H2SO4 30毫升,氯仿200毫升,水浴回流一小时,滤出浸液,药渣再加20%H2SO4 30毫升,氯仿200毫升,水浴回流45分钟,滤出浸液,合并所得浸液,回收氯仿至剩余氯仿约l00毫升,用蒸馏水洗至pH6。
2、大黄酸的分离:上得氯仿液用5%NaHCO3溶液萃取四次,(80、60、40、40毫升),合并NaHCO3液,用盐酸中和至不再析沉淀,析出棕黄色沉淀(若不出现沉淀,水浴上加热)抽滤,水洗70℃烘干,用少量冰醋酸重结晶,析出黄色针晶为大黄酸,抽滤烘干,称重。
3、大黄素的分离:经NaHCO3溶液提取过的氯仿液,继用5%Na2CO3溶液萃取四次(80、60、40、40毫升)。合并Na2CO3液,用盐酸中和至不再析出沉淀,析出棕黄色沉淀(若不出现沉淀,水浴上加热)抽滤,水洗,70℃烘干,用少量无水乙醇重结晶,析出橙色针晶为大黄素、抽滤,烘干,称重。
4、芦荟大黄素,大黄素甲醚和大黄酚混合物的分离:经Na2CO3提取过的氯仿液,再用5%NaOH溶液提取四次(80、60、40、40毫升),合并NaOH液,用盐酸中和至不再析出沉淀,析出黄色沉淀,抽滤,水洗,70℃烘干,称重。
5、芦荟大黄素的分离:4得沉淀物溶于少量氯仿(约30毫升),用0.25%NaOH溶液提取三次(20、10、10毫升),合并提取液用盐酸中和至中性,析出棕黄色沉淀,抽滤,70℃烘干,用少量乙酸乙酯重结晶,析出棕黄色针晶为芦荟大黄素,抽滤,烘干,称重。
6、大黄素甲醚和大黄酚混合物的分离:经0.25%NaOH提取过的氯仿液再用5%NaOH提取三次(20、10、10毫升),合并提取液,用盐酸中和至中性,析出黄色沉淀,抽滤。水洗,70℃烘干,用少量乙酸乙酯重结晶,得大黄素甲醚和大黄酚混合物,抽滤,烘干,称重。
(二)颜色反应及色谱检查
l、取以上各产物少量,分别于试管中,加5%NaOH溶液数滴,观察颜色变化。
2、取以上各产物少量,分别于试管中加浓H2SO4数滴,观察颜色变化。
3、取以上各产物少量,分别于试管中,加少量甲醇溶解,再滴加醋酸镁的甲醇溶液数滴,观察颜色变化。
4、薄层色谱:
吸附剂:硅胶G板,105℃,活化二小时。
展开剂:氯仿—乙酸乙酯—醋酸(4:1:0.2)
检品:各产物的乙醇液
显色:可见光下观察色斑,紫外灯下观察萤光斑点。
四、实验说明及注意事项
1、大黄中蒽醌的存在形式以结合状态为主,游离状态的仅占小部分,为了提高游离蒽醌的得率,在提取过程中采取酸水解和萃取相结合的方法。
2、两相萃取时,不可猛力振摇,只能轻轻旋转摇动,时间可长一些,以免造成严重乳化现象而影响分层,氯仿液用水洗时,尤其易乳化,可加入氯化钠盐析,使两层分离。
五、参考文献
1、林启寿:中草药成分化学,1977,195
2、中药志:上册,27页,1977
3、苏州市中医院,中草药通讯,1977,5(15)
4、何丽一等,药学学报,1980、19(555)
实验五 芦丁的提取与鉴定
芦丁(Rutin)亦称芸香甙(Rutinoside),广泛存在于植物界中,其中以槐花米(为槐树Sophora japnicaL.的花蕾)和乔麦叶中含量较高,本实验以槐花米为原料提取芦丁,其含量为12—16%。
芦丁为维生素P类药物,有助于保护毛细血管的正常弹性,临床上主要作防治高血压的辅助药物,还有调整毛细管壁的渗透作用,临床上作毛细血管性止血药,此外,对于放射性伤害所引起的出血症也有一定治疗作用。
一、实验目的
l、以芦丁为例学习黄酮类化合物的提取方法;
2、掌握黄酮类成分的主要性质及黄酮甙,甙元和糖部分的鉴定方法。
二、实验原理
槐花米中的已知成分
l、芦丁(Rutin)
mpl77—178℃
淡黄色小针状结晶
溶解度:冷水1:8000 热水1:200
冷乙醇1:300 热乙醇1:30
难溶于乙酸乙酯、丙酮,不溶于苯、氯仿、乙醚、及石油醚等溶剂
2、槲皮素(Quercetin)
mp 316℃,313—14℃
(含2分子结晶水) 即芸香甙元,黄色结晶
溶于热乙醇(1:60),冷乙醇(1:650),可溶于甲醇、冰醋酸、乙酸乙酯、丙酮、吡啶,不溶于石油醚、乙醚、氯仿和水中,实验室以稀硫酸水解,乙醇重结晶而制得。
3、还含有皂甙,其粗制品为白色粉末,经酸水解后得二种皂甙元及糖部分,这两种皂甙元是白桦酯醇(Betulin)和槐花米双醇(Sophoradiol),另外还含槐花米甲素、槐花米乙素,槐花米丙素等。
本实验是利用芦丁在热水中和冷水中溶解度相差较大的性质,用热水提取,然后再利用其在热乙醇和冷乙醇中溶解度的差异进行精制。
三、实验内容
(一)芦丁的提取和精制
l、芦丁的提取:取20克槐米于烧杯中用水漂洗于净,捞取上浮的花蕾弃去下沉的杂质,将洗净的花蕾置于1000毫升烧杯中,加沸水500毫升煮沸45分钟,不断补充蒸发掉的水,趁热过滤(用棉花)再用200毫升提取30分钟,合并两次滤液,浓缩1/2体积放置过夜,析出芦丁,抽滤、沉淀用少量冷水洗三次,抽干,60℃以下干燥,称重,计算得率。
2、重结晶:将粗制芦丁研细,加95%乙醇回流溶解(每2克芦丁需加70毫升左右乙醇)趁热过滤,取1/2滤液浓缩至原体积的1/3一1/4,放置,析出结晶,抽滤,干燥称重,得精制芦丁,计算得率。
3、芦丁的水解:取另l/2量芦丁滤液,加入lM硫酸(80毫升)水浴回流l小时(用聚酰胺薄层检查水解是否完全)待全部水解后,蒸去乙醇,冷却、析出槲皮素、抽滤、滤液内含糖。椒皮素沉淀经水洗、抽干、干燥称重,计算得率,再用乙醇重结晶一次,得黄色针晶,为槲皮素精品。
含糖的溶液,取20毫升于水浴上加热,同时于搅拌下加硫酸钡细粉中和至中性。滤除白色的BaSO4沉淀,滤液在水浴上浓缩至1—2毫升,供纸色谱用。
(二)鉴定
l、性质试验
(1)盐酸—镁粉反应,取芦丁少量,加乙醇数滴溶解加浓盐酸5滴,再加少量镁粉,观察颜色变化。
(2)醋酸铅沉淀反应,取芦丁少许溶于热水,加醋酸铅试剂数滴,观察结果。
(3)Molish反应:分别取芦丁和槲皮素少量,置2支试管中加乙醇数滴,加α—萘酚几滴振摇使溶,倾斜试管,沿管壁滴加浓硫酸5滴,静置,观察二层溶液界面处颜色变化,并比较芦丁和槲皮素的区别。
2、色谱鉴定
(1)槲皮素和芦丁的纸色谱鉴定
点样:取新华1号滤纸;距下端3厘米处用铅笔划线,为起始线,隔1.5厘米处点样品。将点好样品的滤纸挂在层析缸中饱和半小时,再上行展开。
a芦丁乙醇液 b芦丁标准品乙醇液 c精制槲皮素乙醇液。
展开剂:正丁醇—醋酸—水(4:1:5,上层)
显色:a可见光下观察色斑,紫外灯下观察荧光斑点。
b喷1%AlCl3乙醇液,再观察荧光斑点。
(2)糖的纸层析鉴定
点样:取新华l号滤纸,同上法点样。
a水解液 b标准葡萄糖水溶液 c标准鼠李糖水溶液
展开剂:正丁醇—醋酸—水(4:1:5,上层)
显色:氨性AgN03溶液(临用时等量0.1N AgN03与10%NH4OH液混合而成)喷洒,加热观察色斑。
槲皮素和芦丁的薄层鉴定
固定相:聚酰胺薄层板
样品:a精制芦丁 b芦丁标准品 c精制槲皮素
展开剂:氯仿—甲醇—丁酮—乙酰丙酮:(16:10:5:1)
显色:a可见光下观察色斑,再于紫外灯下观察荧光斑点
b喷洒AlCL3乙醇液后观察。
四、实验说明及注意事项
芦丁的提取方法较多,如碱提取酸沉淀法,是利用其具有酸性易溶于碱水,加酸酸化后又析出沉淀的性质进行的。
五、参考文献
1、上海药物研究所:中草药有效成分的提取和分离 234—240页
2、中国医学科学院药物研究所:中草药有效成分的研究(第一分册)219—
225 1972
3、日本药学杂志,72(333) 1952;76(1210) 1956,80(304) 1960;80(698) 1960
4、 Chem, Pharm. Bull, ll (167), 1963
实验六 甘草酸的提取
甘草酸(Glycyrrhizic acid)或称甘草皂甙(G1ycyrrhizin)是甘草(G1ycyrrhiza uralensis Fisch.)的根及根茎和光甘草(G.glabra L)的根及根茎中的主要成分,也是有效成分,在甘草中的含量约7一10%味极甜故又称甘草甜素。
甘草是常用和重要的中药之一,有较强的解毒作用,中药用于清热解毒调和诸药,此外尚有类皮质激素、抗炎、抗胃溃疡、镇咳祛痰、解痉等方面的药理作用,甘草中还含有多种黄酮成分,甘草素(Liguiritigenin),异甘草素(Isoliguiritigenin),甘草甙(1iguritin),新甘草甙(Neo1iguritin)新异甘草甙 (Neoisoliguritin)和异甘草素—4—β—葡萄糖—β—洋芜荽糖甙等。
一、实验目的
1、掌握甘草酸的提取原理和方法
2、熟悉皂甙的性质和鉴定方法。
二、实验原理
甘草酸(G1ycyrrhizic acid)
由冰醋酸中结晶出来的为白色柱状结晶,mpl70℃.
易溶于热水、热稀乙醇、丙酮、不溶于乙醇、乙醚等。
在加热、加压及稀酸作用下,可水解为甘草次酸及二分子葡萄糖醛酸。
甘草酸的提取精制原理是:甘草酸在原料中以钾盐或钙盐形式存在,其盐易溶于水,因此用水温浸,提出甘草酸盐,再加硫酸,因难溶于酸性冷水,而析出游离的甘草酸。
甘草酸可溶于丙酮中,加氢氧化钾后,生成甘草酸三钾盐结晶,此结晶极易吸潮不便保存,加冰醋酸后,转变为甘草酸单钾盐,具有完好的晶形,易于保存。
三、实验内容
(一)甘草酸的提取
取甘草粗粉20克,加水150毫升,于水浴上温浸30分钟,棉花过滤,药渣再用100毫升水温浸30分钟,棉花过滤,合并滤液,水浴浓缩至40毫升,滤除沉淀物,放冷加入浓H2SO4并不断搅拌,至不再析出甘草酸沉淀为止,放置,倾出上清液,下层棕色粘性沉淀用水洗涤四次,室温放置干燥,磨成细粉,为甘草酸粗品。
将粗制甘草酸置园底烧瓶中,用50毫升乙醇回流1小时,过滤,残渣再用30亳升乙醇回流30分钟,过滤,合并滤液,浓缩至20毫升,放冷,在搅拌下加入20%KOH乙醇溶液至不再析出沉淀,此时溶液pH=8静置,抽滤,沉淀为甘草酸三钾盐结晶,于干燥器内干燥,称重。
甘草酸三钾盐置小烧杯中,加15毫升冰醋酸,水浴上加热溶解,热过滤,再用少量热冰醋酸淋洗滤纸上吸附的甘草酸,滤液放冷后,有白色的结晶析出,抽滤,用无水乙醇洗涤,得乳白色甘草酸单钾盐。
(二)性质实验及色谱检查
l、泡沫实验
取甘草酸单钾盐水溶液2毫升,置试管中用力振摇,放置10分钟后观察泡沫。
2、醋酐—浓流酸反应(Liebermann—Barchard反应)
取甘草酸单钾盐少量,置白瓷板上,加醋酐2—3滴使溶解,再加半滴浓硫酸观察颜色变化。
3、氯仿—浓硫酸反应
取甘草酸单钾盐少量,加l毫升氯仿,再沿试管壁滴加浓硫酸1毫升,观察两层的颜色变化及荧光。
4、薄层色谱
吸附剂:硅胶G板100℃活化半小时。
展开剂:正丁醇—醋酸—水(6:1:3上层)
样品:甘草酸单钾盐标准品,甘草酸单钾盐70%乙醇液。
显色剂:磷钼酸
四、实验说明及注意事项
1、甘草酸三钾盐极易吸潮,因此必须在干燥器中保存。
2、薄层鉴定中显色前,薄层板上的展开剂需挥干。
五、参考文献
l、中药大辞典,1977(568)
2、《中草药有效成分的研究》中国医学科学院药物研究所1972(23l)
3、《药学通报》1982,1(22)
4、P1anta medica 36(1),74 1979
5、国外医学参考资料,药学分册 (1):60,1978。
实验七 烈香杜鹃挥发油的含量测定
烈香杜鹃系杜鹃花科杜鹃属植物(Rhododendron anthopogonoides Maxim)的叶。近年来用于治疗气管炎,镇咳祛痰都有一定疗效。挥发油为其有效成分之一。
一、实验目的
通过本实验掌握测定挥发油含量的方法,并了解挥发油的一般检识方法。
一、实验原理
烈香杜鹃挥发油中的已知成分。
烈香杜鹃主要含黄酮化合物及挥发油。从挥发油中已分离鉴定出八个单位其中4—苯基丁酮—2含量较高,约占油的40%左右,药理临床实验证明有镇咳平喘作用,此外还有杜鹃酮,松樟脑,香叶烯(月桂烯),柠檬烯,杜鹃烯,10—型芹子烯和γ—芹子烯等。
4-苯基丁酮-α(4-Phenglbufan-2-one)bp 100.5-102℃/8mmHg
杜鹃酮(Germacrone)mp 48℃
松樟脑(Juniper Camphor)mp 155-150℃
香叶烯(月桂烯)(Myrcene)bp.171℃
柠檬烯
杜鹃烯 (limonene)
bp l80—182℃/755mmHg
γ—芹子烯
(γ一Selinene)
本实验是根据植物原料中的挥发油经水蒸汽蒸馏,随水蒸汽馏出与水不相混溶的性质,用水蒸汽蒸馏法提取。
三、实验内容
(一)挥发油测定方法
称取烈香杜鹃粗粉50克,置1000毫升园底烧瓶中加水300毫升,振摇混合后;连接挥发油测定器,并自冷凝管向测定器的刻度部分添加蒸馏水到溢流入烧瓶为止,缓缓加热到沸腾,并保持微沸约5小时,至测定器中的油量不再增加,停止加热,放置片刻,打开测定器下端的活塞,将水缓缓放出,至油层上端到0度线约5毫升处为止,放置一小时,再开启活塞使油层下降至恰与0度线平齐,读取挥发油量并换算成样品的百分含量。
(二)挥发油的性质反应
1.挥发油和油脂滴在滤纸上,加热,观察现象
2.将挥发油滴一滴在滤纸上,喷以新配制的5%香草醛硫酸溶液,观察有何现象。
(三)薄层色谱
吸附剂:硅胶(150—200目),干法铺板。
展开剂:石油醚(60—90℃)——乙酸乙酯(7:3)
样品:挥发油
显色剂:5%香草醛浓硫酸溶液喷洒。
四、参考文献
1.兰州医学院药理教研室:中华医学杂志。
2.兰州医学院训练部,中西医结合资料汇编,1,1974.10
3.中国医学科学院药物研究所,兰州医学院化学研究组汇编,全国防治慢性气管炎杜鹃类药物有效成分黄酮类化合物资料汇编(内部资料)1977.5
(挥发油测定装置)
实验八 丹皮酚的提取、分离和制剂鉴别
牡丹皮为毛莨科植物牡丹Paeonia suffruticosa Andr.的干燥根皮。本品具有清热凉血,活血化瘀的作用,用于温毒发斑,吐血,夜热早凉,无汗骨蒸,经闭痛经,肿痛疮毒,跌打损伤等症。其主要成分有:丹皮酚 (含量l.9%一2%)、牡丹酚甙(丹皮酚甙)、牡丹酚原甙(丹皮酚原甙 (含量5%一6%)、牡丹酚新甙、芍药甙。尚含挥发油约0.15%—0.4%及植物甾醇等。除此外,含丹皮酚的植物还有矮牡丹Paeonia suffruticosa Andr.var spontanea Rehd.的根皮,紫斑牡丹P.papaveracea Andr.的根皮,黄丹皮P.potanini Kom,四川牡丹P.szechuanica Fang,徐长卿Cynanchum pariculatum(Bge)Kitag。
一、丹皮中主要成分的物理性质
l、丹皮酚 (paeonol):C9H10O3,白色针状结晶,mp49.5℃—50.5℃,稍溶于水,具有挥发性,能随水蒸气蒸馏,能溶于乙醇、乙醚、丙酮、氯仿、苯等。UVλ EtOH(max)nm(1g ε):29l(4.01),274(4.17),316(3.84)
2、丹皮酚甙(paeonoside):C15H20O8,无色柱状结晶(乙醇),mp8l℃—82℃,[α]20为—39.33°(c=6.0,H20),可溶于水、醇、丙酮、乙酸乙酯,微溶于氯仿、苯等。
3、丹皮酚原甙(paeonolide):C20H28O12,无色柱状结晶(乙醇—乙酸乙酯),mpl57℃—158℃,〔α〕20为18.20。(c=0.36,H20),可溶于水、醇、丙酮、乙酸乙酯,难溶于苯、石油醚等。
丹皮酚 R=H
丹皮酚甙 R=葡萄糖
丹皮酚原甙 R=葡萄糖—阿拉伯糖
二、目的要求
l、掌握用水蒸气蒸馏法提取丹皮酚的方法。
2、掌握丹皮酚的色谱检识和定性检识。
3、熟悉杞菊地黄丸中主要成分丹皮酚的鉴别方法。
三、基本原理
1、丹皮酚具有挥发性,可随水蒸气蒸馏,又因在冷水中难溶故放冷后析
出结晶。
2、杞菊地黄丸中含有8味中药,牡丹皮中的丹皮酚为有效成分之一,利用丹皮酚溶于乙醚的性质,用乙醚提取,并将醚提取液用酚类的显色反应检识,与丹皮药材提取液和丹皮酚标准品对照鉴别。
四、操作
(一)丹皮酚的提取分离
取市售丹皮①150g,粉碎,加入700m1蒸馏水、10ml乙醇②和40克氯化钠③,浸润后,进行水蒸气蒸馏,收集蒸馏液约300ml,将蒸馏液放冷,静置过夜,有白色针状结晶析出,滤取结晶,干燥,称重。如结晶不纯,可加入95%乙醇至全部溶解(约为粗晶的15倍),抽滤,滤液中加入4倍量的蒸馏水,使溶液呈乳白色,静置后则有大量白色针状结晶析出,若在提取过程中得不到白色结晶,只有油珠状物质沉出,可在蒸馏液中加入少量晶种,摩擦瓶壁后,即有较大量的丹皮酚结晶析出。也可用乙醚萃取蒸馏液几次,合并萃取液后,加无水硫酸钠脱水,回收乙醚至少量,放置析晶,抽滤,结晶用少量水洗2—3次,置干燥器中干燥后称重。
(二)丹皮酚的鉴定
l、显色反应
(1)三氯化铁反应 取丹皮酚结晶少许,滴加5%三氯化铁醇溶液,观察现象。
(2)与浓硝酸反应 取丹皮酚结晶少许,滴加浓硝酸数滴,观察现象。
2、薄层色谱鉴别
薄层析:硅胶G—CMC—Na板
点 样:丹皮酚供试品和对照品的乙醇溶液
展开剂:环己烷—乙酸乙酯(3:1)
展开方式;上行展开,展距10cm。
显 色:喷以盐酸酸化的5%三氯化铁醇溶液,热风吹至斑点显色清晰。
观察记录:记录图谱及斑点颜色。
(三)制剂的薄层色谱鉴别——杞菊地黄丸中丹皮酚的鉴别
本品为蜜丸或水蜜丸,具有滋肾养肝的功效,由枸杞子、菊花、熟地黄、山茱萸(制)、牡丹皮、山药、茯苓、泽泻八味中药组成。
l、预处理
(1)供试液的制备 取本品大蜜丸9g(水蜜丸6g),切碎,加硅藻土4g研匀(水蜜丸研碎),加乙醚40m1,水溶加热回流1h,过滤,滤液挥去乙醚,残渣加乙醇lml使之溶解,即为供试液。
(2)对照液的制备 自制丹皮酚及丹皮酚对照品加乙醇,各制成1m1含1mg丹皮酚的溶液。
2、薄层色谱鉴别
薄层析:硅胶G—CMC—Na板
点 样:供试液、自制丹皮酚及丹皮酚对照品的乙醇液各点样10μl。
展开剂:环乙烷——乙酸乙酯(3:1)。
展开方式:上行展开,展距10cm。
显色:喷以盐酸酸化的5%三氯化铁醇溶液,热风吹至斑点显色清晰。
观察记录:记录图谱及斑点颜色。
五、附注
①丹皮因产地、采收季节的不同,丹皮酚含量差异较大,春秋季节采收含量高,以四川产的含量较高,实验时可以根据含量加减提取的药材量。
②丹皮酚易溶于热水而难溶于冷水,若采用一般装置,由于初馏液中的丹皮酚浓度过大,遇冷易析出结晶,固着于冷凝管内壁,加入乙醇可把固着于冷凝管内壁的丹皮酚溶解而流人接收瓶中。
③加入氯化钠可明显提高蒸馏速度,缩短摄取时间。
六、思考题
1、丹皮酚还可用什么方法提取分离?
2、水蒸气蒸馏法适用于提取什么样的成分?操作中应注意哪些问题?
3、如何利用原药材鉴定成药中的某一成分?
[参考文献]
1、黄泰康 主编,常用中药成分与药理手册,北京:中国医药科技出版社,1994.1040页.
2、刈米达夫等,配糖体の研究一牡丹皮の成份 1.药学杂志(日), 76(8) 1956:917.
3、于津等,牡丹芍药中活性成分的动态研究,药学学报,20(10)1985:782页。
4、张伯祟等,直接蒸馏一紫外分光法测定牡丹皮中的丹皮酚,中成药研究,(7)1983:33页.
5、中华人民共和国药典委员会 编.中华人民共和国药典.(一九九五年版一部).广州:广东科技出版社,1995:147页,497页.
6、徐礼莱:中草药有效成分分析法.上册.北京:人民卫生出版社,1982,l13页.
实验九 预 试 验
一、实验目的
对未知成分的中草药,在分离其有效成分之前,作一初步检查,籍及了解其中所含成分的概貌是很有必要的。
本实验对未知成分的中草药进行一次初步的分离,比较各部分的得量,并按其可能存在的成分选择适当的鉴别反应。进行试验,必要时可以进行纸层析或薄层层析鉴定,最后作出合理的判断。
二、实验原理
预实验主要有两类方法:一类是单项试验法即根据工作需要,有重点地检查其某类成分;另一类是系统预试法,即对中草药中各类成分进行比较全面的定性检查。
系统预试方法很多,常采用的为递增极性溶剂法,就是根据中草药成分亲脂性强弱,选用各种极性不同的溶剂,依次进行提取,使分为若干部分,并按其可能存在的成分选择适当的鉴别反应。最后作出较合理的判断。如将中草药原料依次用石油醚、乙醚、乙醇、水等溶剂进行提取,化学成分依亲脂性强弱依次被提取出来,或者先用甲醇、丙酮等弱亲脂性溶剂提出绝大多数成分,然后用酸性水溶液温浸提出多糖、蛋白质等,总提取物再进一步用溶剂提取,分出酸性、碱性、中性成分、水蒸汽蒸馏分出挥发性成分,本实验就采用后一种方法。
三、实验内容
1、查阅资料(根据所给样品课外进行)
2、初步分离
(1)称取生药粗粉6克(干)或12克(鲜),加丙酮60m1回流40分钟,过滤,再以40m1丙酮回流提取一次,合并滤液,分出l/6浓缩后作部分I。
(2)丙酮浸过的残渣用25ml2%醋酸温浸30分钟过滤,滤液与经浓缩的5/6丙酮提取液合并,以1:1乙醚一氯仿液15ml,10m1×2萃取三次,分成乙醚一氯仿液部分和酸水母液部分。
(3)乙醚——氯仿液以2%Na2CO3液20ml萃取二次,此乙醚一氯仿液浓缩得部分Ⅱ。
i)部分Ⅱ,进行水蒸汽蒸馏,收集馏出液约50m1,以乙醚萃取2次(每次用15m1)乙醚萃取液经脱水,浓缩得部分Ⅱa。
ii)残留液,同样用乙醚萃取2次(每次15ml)醚液经脱水,浓缩后得部分Ⅱb。
(4)Na2CO3水溶液加盐酸酸化至pH3—4,用乙醚萃取2次(每次20m1)醚液经脱水,浓缩得部分Ⅲ。
(5)(2)项上的酸水母液,加氨水碱化至PH9-10,再用1:1乙醚—氯仿液10ml萃取2次,有机部分经脱水,浓缩得部分Ⅳ。氨性水部分加盐酸中和后,浓缩得部分Ⅴ。
(三)各类成分的鉴定
l、挥发油和油脂
(1)油斑试验:将试液滴于滤纸上,能自然挥发或加热后挥发者可能为挥发油。
如果出现持久性的透明斑点,可能为油脂。
(2)香草醛浓HCI试验,将试液滴于滤纸上,喷洒试剂如显紫、兰、黄、红色可能含挥发油。
(对某些酚类、萜类、甾体等皆可显色)
(3)丙烯醛试验:将试液3滴和倍量无水硫酸钠固体置于试管中,直火加热,甘油和甘油脂类能生成有刺激臭味的丙烯醛。(可用斐林试剂检查)
2、蒽醌类
(4)碱液试验(Borntragers 反应):取试液lml加1%NaoH溶液lml,即呈红一红紫色,亦有呈兰色者,表示可能有羟基蒽醌。
(5)醋酸镁试验:取试液0.5ml,加人试剂2—3滴,若有羟基蒽醌类,则会出现橙、兰、紫色等。颜色随羟基数目、位置而定。
3、香豆素
(6)荧光试验:羟基香豆素类的极稀水溶液发生兰色荧光,加氨后呈黄色荧光。
(7)异羟肟酸铁反应:取1N盐酸羟胺甲醇液0.5m1,置于小试管中,加试液数滴,加2N氢氧化钾甲醇液使溶液呈碱性,在水浴上煮沸2分钟,冷却后滴加5%HCI使溶液呈酸性,加1%FeCL3溶液l—2滴,若出现紫红色,表现有香豆素或其他酯类,内酯化合物。
(3)取试品的乙醇液2m1,加1%NaOH液1m1,于沸水浴上加热10分钟(若有
沉淀过滤除去),于澄明液中加2%HCI液酸化后,溶液变混浊,为内酯、香豆素类反应。
[注]可同时取醇浸液2m1,不加试剂对照观察。
4、黄酮类
(9)盐酸镁粉反应:试品的乙醇溶液,加人浓盐酸5滴及少量镁粉,在沸水浴上加热l—2分钟,如呈现红色,表明含有游离黄酮类化合物,如不加镁粉只加浓盐酸即显红色者,可能为花青素。
[注]多数黄酮醇、二氢黄酮、二氢黄酮醇显橙色——紫红,黄酮甙及黄酮醇甙反应不明显,查耳酮、橙酮及儿茶素类无反应。
(10)铝盐络合反应:取试样甲醇液0.5m1,滴加1%AICI3甲醇溶液,呈深黄色,放置后出现黄色荧光者为3,5一位游离羧基或邻二羟基黄酮类。
(11)氨熏试验:将滴有试液的滤纸,加上一滴氨水,立即置紫外灯下观察,有极明显的黄色荧光斑点。
5、糖、低聚糖和甙类
(12)Molish反应:取供试液1m1加10%α一萘酚1—2滴,振摇,倾斜试管,沿管壁加人浓硫酸1m1界面出现紫红色环,表示含糖甙类。
(13)斐林反应:取试品水溶液1-2m1,加入碱性酒石酸铜试剂lml,沸水浴上加热2—3分钟,产生棕红或砖红色沉淀(氧化亚铜),表示含还原糖。
试液与10%硫酸煮沸5—10分钟,冷后以NaOH液中和,再加斐林试剂1m1沸水浴加热2—3分钟,产生的沉淀比水解前多,表示含多糖和甙。
6.甾体、三萜皂甙
(14)皂甙泡沫试验:取试品的中性或弱碱性热水溶液2ml,用力振摇1分钟,如产生多量泡沫,放置10分钟后泡沫没有显著消失即表明含有皂甙成分。
另取两支试管,各加试品热水溶液1ml,一管内加5%NaOH液2ml溶液。另一管加人5%盐酸溶液2m1,将两试管用力振摇一分钟观察两管出现泡沫情况,如两管的泡沫高度相似,表明为三萜皂甙,如含碱液管比含酸液管的泡沫高达数倍,表明有团体皂甙。
(15)浓硫酸醋酐反应(Liebemann—Burchard反应)
取试品少许置白瓷板上,加入醋酐2—3滴,沿白瓷板加入一微滴(用毛细管加入)浓硫酸,交置面出现红色,渐变为紫一兰一绿色等,最后退色。(三萜皂甙最后变兰,甾体皂甙最后变绿色)
(16)氯仿一浓硫酸试验(Salkowshi反应)
将2m1试品的氯仿液,置于试管中,沿管壁滴加浓流酸2m1,氯仿层出现红色,硫酸层有绿色荧光。(如试品不是氯仿溶液,则需将其蒸干,再加2m1氯仿溶解)。
[注]如泡沫反应明显,里伯曼反应红色不明显,可取糖,多糖及甙的水解
液置分液漏斗中,加等量乙醚振摇提取,分出乙醚液,加无水硫酸钠少量脱水,挥去乙醚,再作里伯曼反应。
7、有机酸
(17)pH试纸检查(pH3以下可能含有机酸)
(18)取试液少许加5%AgN03试剂,出现白色沉淀(在毛细管中作)。
(19)溴酚兰试验:将试液滴于滤纸上,喷洒0.1%溴酚兰的乙醇液立即在兰色背景上显黄色斑色。
8、酚类与鞣质
(20)三氯化铁试验,取中性或酸性液3滴,置试管中,加1%FeCL3溶液1滴,出现兰、绿、紫色,表明可能含有酚类或鞣质(必要时可加热)。
(21)明胶沉淀试验,取供试品水溶液,过滤,加入明胶试液l—2滴,出现混浊或白色沉淀可能有鞣质。
(22)取试液lml,加0.1%盐酸小檗碱溶液2—3滴,如变混浊有沉淀表明可能有鞣质。
(23)于滤纸上滴加试液,用三氯化铁——铁氰化钾试剂喷洒,有明显兰色,表明有酚类存在。
9、生物碱
(24)试品酸性水溶液加碘化铋钾试剂产生棕色沉淀或混浊为阳性反应。
(25)试品酸性水溶液,加碘一碘化钾试剂产生橙红色沉淀或混浊为阳性反应。
(26)试品中性水溶液与苦味酸试剂作用产生黄色沉淀,或混浊为阳性反应。
(27)试品酸性水溶液加磷钨酸试剂产生白色沉淀或混浊为阳性反应。
10、强心甙
(28)亚硝酰铁氰化钠反应(1egal反应):将试品溶于2—3滴吡啶中,加入0.3%亚硝酰铁氰化钠溶液1—2滴,再滴加10%NaOH溶液呈红色,渐渐消退。
(29)3、5—二硝基苯甲酸试验(Kedde氏反应),将试品少许加乙醇数滴溶解,加入Kedde试剂,呈紫色。
注:(28)、(29)为五元不饱和内酯环反应。
三氯化铁——冰醋酸反应 (Keller--Kiliani反应)
取试液lml加0.5%FeCl3,醋酸溶液1m1,沿管壁滴加H2SO41m1,二液面间出现棕色环(或其他颜色),冰醋酸层呈绿色→兰色。
(2—去氧糖反应,杂质多时不明显,最好分离纯化后再作)
注:甾体母核反应见(15)、(16)。
11、蛋白质、多肽及氨基酸
(31)双缩尿 (Biuret)试验:取试样0.5m1,加入1%NaOH溶液l—2滴,滴加0.5%CuSO4试液2滴,摇匀,出现紫色,红紫色表明含多肽或蛋白质。
(32)茚三酮(Ninhydrin)试验:取试液0.5m1,加入试剂l—2滴摇匀,在沸水浴上加热数分钟,应出现兰色,紫色或红紫色,或将试液滴于滤纸上,烤干,喷洒试剂,再于100℃加热。2—5分钟呈色亦可。
[参考文献]
l、《中草药化学》:全国中专卫生学校试用教材。1979 188—192
2、中国医学科学院药物研究所72年;中草药有效成分的研究。
附录二 常用溶剂的回收及精制方法
在我们的实验中,常常需要应用很多的有机溶剂,这些溶剂用过以后就会混入许多有机及无机物质,并带进了很多水分,除去这些杂质和水分后,这些溶剂就又可以重新使用了,因此,再生溶剂也是贯彻增产节约的具体表现,在分析和色谱实验中对溶剂的纯度要求更高。一般重蒸的溶剂或市售工业品均不可直接应用,必须进一步精制,否则将影响实验的结果。因此,将各种溶剂的再生和精制方法分述于下:
一、石油醚
石油醚是石油馏分之一,主要是饱和脂肪烃的混合物,极性很低,不溶于水,不能和甲醇、乙醇等溶剂无限止地混合,实验室中常用的石油馏分根据沸点不同有下列数种,其再生方法大致相同。
沸 点 比 重
轻石油醚 35—60℃ 0.59—0.62
重石油醚 60一80℃ 0.64一0.66
汽 油 80一120℃ 0.67一0.72
汽 油 120—150℃ 0.72 —0.75
再生方法:用过的石油醚,如含有少量低分子醇,丙酮或乙醚,则置分液漏斗中用水洗数次,以氯化钙脱水、重蒸、收集一定沸点范围内的部分,如含有少量氯仿,在分液漏斗中先用稀碱液洗涤,再用水洗数次,氯化钙脱水后重蒸。
精制方法:工业规格的石油醚用浓硫酸,每公斤加50一100克振摇后放置一小时,分去下层硫酸液,可以溶去不饱和烃类,根据硫酸层的颜色深浅,酌情用硫酸振摇萃取二、三次。上层石油醚再用5%稀碱液洗一次,然后用水洗数次,氯化钙脱水后重蒸,如需绝对无水的,再加金属钠丝或五氯化二磷脱水干燥。
二、环乙烷
沸点81℃,性质与石油醚相似,再生时先用稀碱洗涤。再用水洗,脱水重蒸。其精制方法将工业规格环乙烷加浓硫酸及少量硝酸钾放置数小时后,分去硫酸层,再以水洗,重蒸,如需绝对无水的,再用金属钠丝脱水干燥。
三、苯
沸点80℃,比重0.879,不溶于水,可与乙醚、氯仿、丙酮等在各种比例下混溶,纯苯在54℃时固化为结晶,常利用此法纯化。
再生方法:用稀碱水和水洗涤后,氯化钙脱水重蒸。
精制方法:工业规模的苯常含有噻吩、吡啶和高沸点同系物如甲苯等,可将苯1000毫升,在室温下用浓硫酸每次80毫升振摇数次,至硫酸层呈色较浅时为止,再经水洗,氯化钙脱水重蒸,收集79—8l℃馏分。对于甲苯等高沸点同系
物,则用二次冷却结晶法除去,苯在54℃固化成为结晶,可以冷却到0℃,滤取结晶,杂质在液体中。
四、氯仿
沸点6l℃,比重1.488,不溶于水,易与乙醚、乙醇等混溶,在日光下易氧化分解成Cl2、HCl、C02及光气(COCl2),后者有毒,故应贮在棕色瓶中。氯仿在稀碱水作用下易分解产生甲酸盐,在浓碱水作用下则生成碳酸盐。
再生及精制方法:医用氯仿含有1%酒精作为安定剂,以防止它的分解,可用水洗涤,氯化钙脱水重蒸,收集61℃的馏分,贮于棕色瓶中。
五、四氯化碳
沸点77℃,比重1.589,极性很低,不溶于水;工业规格的四氯化碳中常含有2—3%二硫化碳,其除去方法取1000毫升四氯化碳加5%KOH乙醇溶液100毫升,60℃加热三十分钟,冷却后,用水洗涤(氯化钙或固体)分去水层,再用少量浓硫酸振摇多次,直至硫酸不变色,最后用水洗涤,氯化钙或固体氢氧化钠脱水,加石蜡油少许后蒸馏可得精制品。
(附注)氯仿和四氯化碳脱水干燥时,切忌用金属钠,否则将发生爆炸事故。
六、二硫化碳
沸点46℃性质与四氯化碳相似,纯的二硫化碳为无色液体,味香,有毒性,市售工业规模的常含硫化氢、硫氢化碳等分解产物因而其味难闻。二硫化碳久置色变黄。精制时先用金属汞振摇,再用饱和氯化汞冷溶液振摇,最后再用高锰酸钾液洗涤后蒸馏而得。
七、乙醚
沸点35℃,比重0.714,在水中的溶解度为8.11%。用过的乙醚常含有水及醇,如用水洗涤损失很大,可用饱和氯化钙水液洗涤,同时又可去除乙醇,再以无水氯化钙脱水干燥,重蒸即得。
乙醚久置于空气中,尤其是日光下暴露,则逐渐氧化成醛,酸及过氧化物。当过氧化物达到万分之几时,蒸馏时有发生爆炸的危除,过氧化物的存在可以用碘化钾溶液与少量乙醚共振摇生成游离碘而检出,其除去法可用稀碱液,高锰酸钾液,亚硫酸钠液顺次洗涤,再用水洗,干燥,重蒸而得,贮存时加少量表面洁净的铁丝或铜以防止氧化。
另法除法少量醇类可在乙醚中加少量高锰酸钾粉末和1—2块(10克左右)氢氧化钠,放置数小时后,在氢氧化钠表面如有棕色的醛缩合树脂生成者,重复这一操作直至氢氧化钠表面不生棕色物为止,然后将乙醚倒入另一瓶内,加无水氯化钙脱水,重蒸而得,如须绝对无水的,再将金属钠压成钠丝加入,瓶塞打孔,附一氯化钙管,放置为了减少蒸发,在氯化钙管上安装一根一端拉成毛细管的玻璃管以与外界相通。
八、丙酮
沸点56℃,比重0.792,与水、醇能任意混溶。
再生方法:丙酮中如含有多量的水时,可加食盐或固体碳酸钾等盐类,盐析成二层,分去下层盐水层,上层丙酮液蒸馏收集54—57℃馏分,再用无水氧化钙脱水干燥重蒸。
精制方法 、
1.一般工业用丙酮,常会有甲醇、醛和有机酸等杂质,精制时加高锰酸钾粉末回流,所加的量应使丙酮一直保持紫色,如不加热,放置3—4天也可,加热后冷却,滤去沉淀,加无水碳酸钾或氯化钙脱水干燥,蒸馏收集。
2.如丙酮中混有少量乙醇、乙醚、氯仿等溶剂,精制时加二倍量的饱和亚硫酸氢钠溶液振摇,生成亚硫酸氢钠丙酮加成体,再在其中加等量的酒精,析出结晶,过滤收集,顺次以酒精、乙醚洗涤,干燥。将此结晶与少量水相混合,加入10%碳酸钠或10%盐酸使加成物分解、滤液分级蒸馏,取丙酮之馏分,再加无水氯化钙或碳酸钾脱水干燥,重蒸而得。
注意:丙醇不宜用金属钠或五氧化二磷脱水。
九、乙醇
沸点78℃,比重0.79与水能任意混溶,蒸馏时与水共沸,共沸点78.1℃,共沸混溶液含水4.43%为95%乙醇。
再生方法:先在用过的乙醇中加生石灰(氧化钙),每立升加25—50克,加热回流脱水后,分级蒸馏,收集76—81℃的馏分,含醇80—90%。再置园底烧瓶中,加计算量多一倍的生碳,回流五小时,再蒸馏收集76—78℃的馏分,可达98.5—99.5%。
如须绝对无水者;可用下列二法之一:
1、99.5%乙醇l000毫升,加27.5克苯二甲酸二乙酯和7克金属钠,放置后蒸馏得无水乙醇
C6H4(COOC2H5)2+2C2H5ONa+2H20→
C6H4(COONa)2+4C2H50H
2、98%以上的乙醇60毫升,置于2立升的园底烧瓶中,加入5克金属镁,0.5克碘,使发生反应促进镁溶解成醇镁,再加900毫升乙醇,回流加热5小时,蒸馏可得100%乙醇。
(C2H5O)2Mg+2H2O→2C2H5OH++Mg(OH)2
如用以紫外光谱分析,要求较高,普通发酵乙醇常混有少量醛和酮,无水乙醇用苯共沸蒸馏所得者常含有苯、甲苯,均不宜于光谱分析用。其精制法如下:95%普通乙醇l000毫升,加人25毫升12(NH4)2SO4,在水浴上迥流加热数小时以除苯及甲苯等杂质。蒸馏,初馏分50毫升及残馏分l00毫升除去。主馏分中加硝酸银8g,加热使溶解,溶解后再加入粒状氢氧化钾15克。迥流加热l小时,此时溶液从粘土色的AgOH悬浊液变为黑色的还原银粒凝集沉淀下来。此反应约需20—30分钟。如果黑色沉淀生成很早,表示能被氧化的物质存在较多。蒸馏后的溶液再以少量硝酸银和氢氧化钾(1:2W/W)加入重复上述操作直至没有黑色沉淀生成为止,再继续加热三十分钟,蒸馏,初馏分约50毫升及残馏分约100毫升除去。主馏分收集,但有可能带入微量的碱和银离子,会促进乙醇的氧化。应重蒸馏一次,由此法制得的乙醇含水3—6%在206nm外透明,200nm处有末端吸收。
十、甲醇
沸点65℃,比重0.79,能与水、乙醇、乙醚、氯仿作任何比例的混溶,不与水共沸,利用分馏法可得99.8%的浓度,绝对无水的甲醇,可用镁和碘的方法制得(同乙醇项下)。甲醇有毒,对视神经有损伤,应用和操作时应注意。
精制方法:工业规模的甲醇中,主要含丙酮和甲醛杂质,可用硫酸汞酸性溶液与甲醇一起加热,使丙酮生成络合物析出,或碘的碱性溶液共热使醛或酮氧化成碘仿,然后再分馏精制。
注意甲醇不能用生石灰脱水,因CaO能吸收20%甲醇,CaO、CH30H、H20为一平衡,完全脱水不可能。
十一、乙酸乙酯
沸点77℃,比重0.90,含水的乙酸乙酯在日光下会逐渐水解为醋酸和乙醇,精制时以5%碳酸钠(或碳酸钾)溶液,饱和氯化钙溶液分别洗去醋酸和醇再以水洗、分级蒸馏取乙酸乙酯馏分,再经无水氯化钙脱水干燥后重蒸一次,或在乙酸乙酯中加少量水,每500克加2克水,蒸馏,水和乙醇在第一馏分中即被蒸出。
十二、醋酸
沸点118℃。冰点16.5℃,比重1.06,纯的醋酸(99—100%)在较低温度时结成固体,故又称冰醋酸,其精制可用冰冻法,即冷却至0一10℃醋酸结成结晶,分去液体,结晶加热复重熔化,再经冷冻一次,可得水醋酸。
醋酸中如含有乙醇和醛等杂质,可在醋酸中加2%左右的重铬酸钾(或钠)后进行分馏,若含有少量水分则加适量的醋酐后进行分馏,收集l17℃一l18℃的馏分。
十三、吡啶
沸点116℃,比重0.98,能与水、乙醇、乙醚等混液,和水共沸,共沸点92—93℃,吡啶中的水分可加适量的固体氢氧化钠放置,分去析出水层后,再加固体氢氧化钠至无水层分出为止,蒸馏,收集l16℃馏分,为无水吡啶。
十四、二甲基甲酰胺(简称DMF)
沸点153℃,比重0.95,能与水、乙醇、乙醚等许多有机溶剂任意混溶。二甲基甲酰胺与水形成共沸混合物,故含有水分的二甲基甲酰胺,不能用分馏法除去,可加无水碳酸钾干燥后,蒸馏精制。
附录三 常用干燥剂性能的说明
化学干燥剂可分二类,一类是与水可以生成水合物的,如硫酸、氯化钙、硫酸铜、硫酸钠、硫酸镁和氯化镁等。另一类与水反应后生成其他化合物的,如五氧化二磷、氧化钙、金属钠、金属镁、金属钙和碳酸钙等,必须注意的是有些化学干燥剂是一种酸或与水作用后变为酸的物质,也有一些化学干燥剂是碱或与水作用后变为碱的物质,在用这些干燥剂时就应考虑到被干燥物的酸碱性质。应用中性盐类作干燥剂时,如氯化钙,它能与多种有机物形成分子复合物,也要加以考虑。因此在选择干燥剂时首先应了解干燥剂和被干燥物的化学性质是否相容,下面介绍一些实验室常用的干燥剂的性能。
一、氯化钙
对固体、液体和气体的干燥均可使用。有干燥能力的是含二分子结晶水的氯化钙CaCl22H2O,潮解吸水后成为含六分子结晶水的氯化钙CaCl26H2O加热至30℃时成CaCl24H2O,至200℃恢复为CaCl22H2O,如加热至800℃则水分完全失去,成为熔融的氯化钙,可以用氯化钙脱水的化合物有烃类、卤代烃类、醚类,对沸点较高的溶剂,干燥后重蒸溶剂时,应将干燥剂滤出,不可一起加热蒸馏,以免被吸去的水分在加热时再度放出,它的缺点是脱水能力不强,并且能和多种有机物生成复合物,如醇、酚、胺、氨基酸、脂肪酸等,因此不可用作为醇等溶剂的脱水于燥剂。
对结构不明的化合物溶液,就不宜使用氯化钙来干燥。
二、硫酸钠
无水硫酸钠可用于中性,酸性和碱性物质的脱水干燥剂,对有机物没有反应,可以广泛应用,吸水后成为带有十分子结晶水的硫酸钠Na2SO4·10H2O,但脱水能力弱而且作用慢,不能用加热来促使脱水,因为含水的硫酸钠在33℃以上又失结晶水,对于含水量较多的醇类不宜用作脱水干燥剂,适用于醚、苯、氯仿等溶剂,新买来的应加热焙干后使用。
三、硫酸镁
性质同硫酸钠,吸水效力强一些,与水生成水合物含七分子结晶水。
四、硫酸铜
制备无水醇时常加以应用,是相当弱的干燥剂。无水硫酸铜浅绿色,生成水合物质变兰CuSO45H2O,根据变兰的反应说明吸水过程在进行,故可用来检验溶剂的无水程度,CuSO45H2O加热至100℃失去四分子结晶水可以由此再生。加热温度不宜增至220——230℃否则就生成碱性盐类失去水合的效力。
五、硫酸钙
无水硫酸钙由石膏加热至160一180℃而得,如在500—700℃灼烧所得的无
水硫酸钙,几乎不能与水结合。它是强烈干燥剂之一,但吸水量不大,只能达到其全重量的6.6%,吸水后形成相当稳定的水合物2CaSO4·H2O,它和其它形成水合物的盐类不同,被干燥的有机液体不需要把它事先分开,可以放在一起蒸馏,甲醇、乙醇、乙醚、丙酮、甲酸和醋酸用硫酸钙脱水可得良好的效果。
六、苛性碱
苛性钠(NaOH)和苛性钾(KOH)是碱性干燥剂,适用于干燥有机碱类,如氨气、胺类、吡啶、重氮甲烷,生物碱等,作为干燥器内的干燥剂,用来排除被干燥物质挥发出来的酸性杂质时,应用更多,苛性钾的效力较苛性钠大60倍,对于酸性物或酮,醛等均不适用。
七、碳酸钾
无水碳酸钾的碱性比苛性碱弱,应用范围较广一些,除适用于碱性物质外,对醇类也适用。
八、氯化钙
俗称生石灰,也是一种碱性干燥剂,实验室常用来制造无水乙醇,因为来源方便,生成氢氧化钙不溶于乙醇,要得到绝对无水的乙醇,需要用过量很多的氧化钙,对1克水要5克块状氧化钙(理论量是3.11克)干燥有机碱液体也可用之,氧化钙不适用于甲醇,因CaO、H2O、CH3OH三者间与形成的复合物成一平衡,不完全脱水,而且要吸收20%的甲醇。
九、金属钠
金属钠有很强的脱水作用,广泛被应用于各种惰性有机溶剂的最后干燥,如用于乙醚、苯、甲苯、石油醚等,由于金属钠有可工塑性,脱水时可将钠块周围的杂质切去,用压钠机压成条状故入置有溶剂的容器内,这样使金属钠与液体接触的表面大大增加,不致由于金属钠含有的杂质在钠块表面形成一层薄膜,妨碍进一步与水作用,必须注意对CHCl3,CCl4及其他含有-OH,>C=O等反应性强的官能团的溶剂都不能用金属钠脱水,含水量多的溶剂也不能用,因为钠遇水发生爆炸,易引起危险事故。
十、浓硫酸
浓硫酸是一种酸性干燥剂,由于它对许多有机化合物的腐蚀性限制了它在干燥上的应用,因此硫酸多半应用于无机物或作为干燥器内的干燥剂,对于气体,并不是所有中性和酸性的气体对硫酸都不起作用,硫酸除了酸的作用外还有氧化作用,例如溴化氢遇到硫酸将大部分被氧化成溴,干燥器内以硫酸为干燥剂的应用很广,但是真空干燥器内应用硫酸应十分小心,因为它在1毫米汞柱的压力下有一部分要挥发,它的蒸汽与干燥物质就能起作用,放在干燥器内的硫酸不需要纯的,在硫酸中可加1%硫酸钡(18克硫酸钡加在一升硫酸内,比重1.84)。当硫酸吸水浓度降低至93%时,即析出BaSO4、2H2SO4·H2O的针状结晶,当硫酸浓度降低至84%时,(H2SO4·H2O)变成很细的结晶,如果我们发现有细小的硫酸钡结晶出现时,就应该换新硫酸。
十二、五氧化二磷
五氧化二磷即是磷酸酐,吸水后生成磷酸;它的脱水反应是不可逆的,在酸性干燥剂中它的效力要算最高,可用于一般固体,气体和惰性液体的脱水。碱性物质或有羟基的化合物不宜用五氧化二磷来脱水。它的最大缺点是吸水后表面生成一层很粘的磷酸妨碍它的进一步的干燥作用,必须注意五氧化二磷中常含有少量的三氧化二磷,此物大量地与热水作用将生成很毒的磷化氢。
2P2O3+6H2O——PH3+3H3PO4
十三、硅胶
二氧化硅与少量水(2—10%)结合形成的胶状硅胶(SiO2XH2O),称为硅胶,呈无色透明玻璃块状,其中有无数目不能见的细孔,籍毛细现象吸收湿气,发挥干燥能力,常用为气体干燥剂,吸水硅胶外观无变化,为了便于观察,可加COCl2盐,干燥时呈兰色,吸水后呈淡黄色(COCl2用量少时则退色)。
再生时将硅胶铺在器皿中成一薄层,故人烘箱150—180℃加热小心勿超过200℃。
下面是各种干燥剂按效力降低的次序排列:
第Ⅰ类 第Ⅱ类 第Ⅲ类
1.P2O5 10.Mg(ClO4)23H2O 16.H2SO4(95%)
2.A12O3 11.CaO 17.CaCl2(工业无水)
3.B2O3 12.CaCl2(无水) 18.CaCl2(颗粒)
4.BaO 13.CaBr2 19.ZnCl2(熔融)
5.Mg(Cl04)2 14.NaOH(熔融) 20.ZnBr2
6.KOH(熔融) 15.Ba(Cl04)2 21.CuSO4
7.H2SO4 22.MgSO4
8.硅胶 23.Na2SO4
9.CaSO4
上述三类干燥剂,每一类在干燥空气时,于25—30℃以l一3升/分的速度通过,结果在干燥空气中残留的水分各为:
第一类(1—9) l×10 -5——1×lO -3毫克/升
第二类(10—15) 1×10 -2——l×10 -1毫克/升
第三类(16—23) l×10 -1——1×10 0毫克/升
仪 器 清 单(每组)
-
天然药物实验报告
天然药物化学实验实验报告实验一槐米中芦丁的提取精制和鉴定一实验目的要求1制备芦丁供药用2通过芦丁的制备掌握黄酮类化合物的提取分离的…
-
天然药物化学实验报告
天然药物化学实验报告1天然药物化学实验报告一实验题目丹皮酚的提取分离及结构鉴定二实验目的通过对该选题进行资料查阅方案设计试验结果分…
- 天然药物化学第四组实验报告
-
天然药物化学实验报告书写格式、版面标准[1]
天然药物化学实验报告书写格式版面标准1天然药物化学实验报告书写格式版面标准一基本格式天然药物化学实验报告的各个组成部分按下述顺序依…
-
天然药物化学实验
天然药物化学实验课程代码课程名称天然药物化学实验学分3学时48其中实验学时48先修课程有机化学天然药物化学一目的与任务目的是促进学…
-
天然药物化学实验报告
天然药物化学实验报告1天然药物化学实验报告一实验题目丹皮酚的提取分离及结构鉴定二实验目的通过对该选题进行资料查阅方案设计试验结果分…
-
20xx年天然药物化学实验讲义
天然药物化学实验讲义实验须知天然药物化学实验教学是天然药物化学课程的重要组成部分是使学生进一步理论联系实际掌握天然药物有效成分提取…
-
天然药物实验报告
天然药物化学实验实验报告实验一槐米中芦丁的提取精制和鉴定一实验目的要求1制备芦丁供药用2通过芦丁的制备掌握黄酮类化合物的提取分离的…
-
天然药物化学实验书-合集-20xx
天然药物化学实验指导循环使用请勿带走目录实验一槐花米中芦丁的提取实验二芦丁水解液的纸色谱鉴定实验三大黄中大黄素的提取实验四大黄中大…
-
天然药物化学实验讲义
天然药物化学实验讲义实验须知实验一薄层板的制备活度测定及应用实验二粉防已生物碱的提取分离与鉴定实验三掌握掌叶防已碱的提取及硫酸延胡…