药物合成综合实验大纲
《药物合成综合实验》课程教学大纲
课程代码:05197490
课程名称:药物合成综合实验
课程学分:3 课程学时:48(实践学时)
课程性质:专业必修 课程归属:科学素养
开课部门:生物工程学院 建议修读学期:20##-20##-1
适用对象:制药工程(本)
实践场所:6301/6302
« 课程概况
n 教学目的
我国的药物研究正经历着一个从仿制到创制新药为主的历史性转变时期,要抓住此次机遇,顺利实现这个历史性转变,关键在于创新型人才的培养。药物合成中的专业技能实训是制药及药学类专业重要的专业课,也是一们实验性很强的课程,在药学类创新型人才培养方面发挥着重要作用。本课程依据药物合成反应教学大纲的要求制定实训内容,目的是通过实验加深理解药物化学的基本理论和基本知识,掌握合成药物的基本方法,掌握对药物进行结构改造与修饰的基本方法,进一步巩固有机化学实验的操作技术及有关理论知识,培养学生理论联系实际的作风,实事求是,严格认真的科学态度与良好的工作习惯。
n 课程地位
先修课程为无机化学、分析化学、有机化学、药物合成反应。学好相关的理论基础知识,有助于对实践进行指导,可以加深课程理论联系实践的教学体会,增加学生对专业技能的掌握。
n 学时分配

« 教学内容及要求
n 1. 二苯甲醇的制备(实践内容)
u 实践目的和要求
了解酮的还原反应机理、还原剂的种类及特点。掌握硼氢化钠还原剂的使用条件。.掌握粗产品提纯方法。
u 实践方法和步骤
在配有搅拌子、温度计、回流冷凝器的 50 mL 的三颈瓶中,加入二苯酮2.0 g、95%的乙醇 20 mL,水浴加热至反应物全溶,冷却至室温,搅拌下慢慢加入硼氢化钠 0.6 g,加入速度以反应温度保持在 50℃以下为宜,加毕,加热反应物至回流反应 1 小时,冷却到室温,加入 10 mL 水稀释反应液,再加入 2 mL 10%稀盐酸分解未反应的硼氢化钠,待冷却到室温后抽滤,水洗滤饼,抽干得粗产品,以石油醚(b.p30-60℃)为溶剂重结晶可制得精品。 计算产率。
思考:除了本实验提供的方法外可否采用其他反应途径制备二苯甲醇。
n 2. 2-亚胺基-4-噻唑酮的制备(实践内容)
u 实践目的和要求
掌握缩合反应机理; .掌握粗产品提纯方法。
u 实践方法和步骤
将 3.8 g 硫脲、30 mL 95%乙醇加到 100 mL 圆底瓶中,水浴加热至回流, 搅拌约 15min,使硫脲全部溶解,滴加 6.2 g 氯乙酸乙酯,约 15~20 min 滴完, 继续回流反应 2.5 h, 冷至室温, 析出大量白色结晶, 抽滤、少量乙醇洗涤、干燥,得产品为白色固体,计算收率。
思考:写出该环合反应的机理
n 3. 2,4-噻唑二酮的制备(实践内容)
u 实践目的和要求
了解亚胺水解制备酮的反应机理;掌握粗产品重结晶提纯方法
u 实践方法和步骤
将 4.4 g 2-亚胺基-4-噻唑酮、45 mL 20%盐酸加到 100 mL 圆底瓶中, 油浴加热回流反应 4 h, 将反应液浓缩,冷却到室温后析出结晶, 抽滤, 用少量冷水洗涤, 得粗产品约4.0 g,用70%乙醇重结晶,得白色粗针状结晶,测熔点。
思考:写出该水解反应的机理
n 4. 3,4-二氯硝基苯的合成(实践内容)
u 实践目的和要求
学习硝化反应,掌握条件与方法; 掌握粗产品提纯方法。
u 实践方法和步骤
在装有搅拌器、温度计的三颈瓶中,先加入硝酸5.1 g,水浴冷却下,用滴管滴加硫酸7.9 g,控制滴加速度,使温度保持在50℃以下。滴加完毕,用滴液漏斗于40~50℃内滴加邻二氯苯3.5 g,5 min内滴完,换上回流冷凝器,升温至60℃,反应2 h,静置分层,吸取上层油状液体入5倍量水中,搅拌,固化,放置30 min,过滤,水洗至pH 6~7,真空干燥,称重,计算收率。
[注意]:
1. 本反应是用混酸硝化。硫酸可以防止副反应的进行,并可以增加被硝化物的溶解度;硝酸生成NO2+,是硝化剂。
2. 此硝化反应需达到40℃才能反应,低于此温度,滴加混酸会导致大量混酸聚集,一旦反应引发,聚集的混酸会使反应温度急剧升高,生成许多副产物,因此滴加混酸时应调节滴加速度,控制反应温度在40~50℃。
[思考题]
1. 配制混酸是能否将浓硝酸加到浓硫酸中去?为什么?
2. 如何检查反应是否已进行完全?
n 5. 桂皮酸的制备(实践内容)
u 实践目的和要求
掌握Perkin反应的基本原理及操作方法
u 实践方法和步骤
 配料比(重量比):苯甲醛∶醋酐∶醋酸钾=1∶1.43∶0.6,在250 mL圆底烧瓶中加入20 g苯甲醛,20ml醋酐和新熔焙过的12 g KOAc。安装空气冷凝器及CaCl2干燥管,在油浴上加热回流振摇使溶解,维持油浴温度160℃(内温约150℃)1.5小时,然后升温至170℃加热2.5小时。(内温约160℃~170℃)。
配料比(重量比):苯甲醛∶醋酐∶醋酸钾=1∶1.43∶0.6,在250 mL圆底烧瓶中加入20 g苯甲醛,20ml醋酐和新熔焙过的12 g KOAc。安装空气冷凝器及CaCl2干燥管,在油浴上加热回流振摇使溶解,维持油浴温度160℃(内温约150℃)1.5小时,然后升温至170℃加热2.5小时。(内温约160℃~170℃)。
反应完成后,取下反应瓶,将反应物倒入125 mL热水中,并用少量水冲洗反应瓶,在反应液中加入适量Na2CO3,调pH至8然后倒入500 mL圆底烧瓶中进行水蒸汽蒸馏,除尽未反应的苯甲醛后,加入适量活性炭约2匙,煮沸15分钟,趁热抽滤,冷却后,慢慢滴加浓盐酸酸化,边加边搅拌,使桂皮酸结晶析出完全,抽滤,水洗涤,干燥得粗品,用稀乙醇(V(水) ∶V(95%EtOH) = 3∶1)重结晶,得桂皮酸结晶,熔点131.5~132 ℃。
思考题
1.桂皮酸合成为什么必须在无水条件下进行?
2.醋酸钾为何必须新鲜熔融,如想提高收率可采取什么措施?
n 6. 烟酸的制备
u 实践目的和要求
掌握高锰酸钾氧化法对芳烃的氧化原理及实验方法;
熟悉酸碱两性有机化合物的分离纯化技术;
了解烟酸的合成路线。
u 实践方法和步骤
1、投料比

2、操作步骤
 在配有回流冷凝管、温度计和搅拌子的三口烧瓶中。加入3-甲基吡啶5 g、蒸馏水200 mL水,水浴加热至85℃。在搅拌下,分批加入高锰酸钾21 g, 控制反应温度在85~90℃,加毕,继续搅拌反应1 h。停止反应,改成常压蒸馏装置,蒸出水及未反应的3-甲基吡啶,至流出液呈现不浑浊为止,约蒸出130 mL水,停止蒸馏,趁热过滤,用12 mL沸水分三次洗涤滤饼(二氧化锰),弃去滤饼, 合并滤液与洗液,得烟酸钾水溶液。
在配有回流冷凝管、温度计和搅拌子的三口烧瓶中。加入3-甲基吡啶5 g、蒸馏水200 mL水,水浴加热至85℃。在搅拌下,分批加入高锰酸钾21 g, 控制反应温度在85~90℃,加毕,继续搅拌反应1 h。停止反应,改成常压蒸馏装置,蒸出水及未反应的3-甲基吡啶,至流出液呈现不浑浊为止,约蒸出130 mL水,停止蒸馏,趁热过滤,用12 mL沸水分三次洗涤滤饼(二氧化锰),弃去滤饼, 合并滤液与洗液,得烟酸钾水溶液。
将烟酸钾水溶液移至500 mL烧杯中,用滴管滴加浓盐酸调pH值至3-4(烟酸的等电点的pH值约3.4,注意:用精密pH试纸检测),冷却析晶,过滤,抽干,得烟酸粗品。
3、精制
将粗品移至250 ml圆底烧瓶中,加粗品5倍量的蒸馏水,水浴加热,轻轻振摇使溶解,稍冷,加活性碳适量,加热至沸腾,脱色10 min,趁热过滤,慢慢冷却析晶(注1),过滤,滤饼用少量冷水洗涤,抽干,干燥,得无色针状结晶烟酸纯品,mp236~239℃。
思考题:
氧化反应若反应完全,反应液呈什么颜色?
为什么加乙醇可以除去剩余的高锰酸钾?
n 7-8, 苯佐卡因的合成
u 实践目的和要求
学习酯化及还原反应,掌握反应条件与方法; 掌握粗产品提纯方法。
u 实践方法和步骤
(1)对硝基苯甲酸乙酯的制备(酯化)
在干燥的100 mL圆底瓶中加入对硝基苯甲酸6 g,无水乙醇24 mL,逐渐加入浓硫酸2 mL,振摇使混合均匀,装上附有氯化钙干燥管的球型冷凝器,油浴加热回流80 min(油浴温度控制在100~120℃)。浓缩反应液(蒸出部分乙醇),稍冷,将反应液倾入到100 mL水中,析出白色沉淀,抽滤;滤渣移至烧杯中,研细,加入5%碳酸钠溶液10 mL(由0.5 g碳酸钠和10 mL水配成),研磨5 min,测pH值(检查反应物是否呈碱性),抽滤,用少量水洗涤,干燥,计算收率。
(2)对氨基苯甲酸乙酯的制备(还原)
在装有搅拌棒及球型冷凝器的250 mL三颈瓶中,加入35 mL水,2.5 mL冰醋酸和已经处理过的铁粉8.6 g,开动搅拌,加热至95~98℃ 反应5 min,稍冷,加入对硝基苯甲酸乙酯6 g和95% 乙醇35 mL,回流反应90 min。稍冷,在搅拌下,分次加入温热的碳酸钠饱和溶液(由碳酸钠3 g和水30 mL配成),搅拌片刻,立即抽滤(布氏漏斗需预热),滤液冷却后析出结晶,抽滤,产品用稀乙醇洗涤,干燥得粗品。
精制
将粗品置于装有球形冷凝器的100 mL圆底瓶中,加入10~15倍(mL/g)50% 乙醇,在水浴上加热溶解。稍冷,加活性碳脱色(活性碳用量视粗品颜色而定),加热回流20 min,趁热抽滤(布氏漏斗、抽滤瓶应预热)。将滤液趁热转移至烧杯中,自然冷却,待结晶完全析出后,抽滤,用少量50% 乙醇洗涤两次,压干,干燥,测熔点,计算收率。
[思考题]
1. 氧化反应完毕,将对硝基苯甲酸从混合物中分离出来的原理是什么?
2. 酯化反应为什么需要无水操作?
3. 铁酸还原反应的机理是什么?
n 9-10. 香豆素-3-羧酸的合成
u 实践目的和要求
1.掌握 Knovengel反应的基本原理和操作方法.
2.熟悉回流和重结晶的操作.
3.巩固Perkin合成法。
u 实践方法和步骤
1.香豆素-3-甲酸乙酯的合成
在干燥的100 mL圆底烧瓶中,加入4.2 mL水杨醛、6.8 mL丙二酸二乙酯、25 mL无水乙醇、0.5 mL六氢吡啶和2滴冰醋酸,放入几滴沸石后,装上回流冷凝管,冷凝管上口接一氯化钙干燥管。在水浴上加热回流2 h。稍冷后将反应物转移到锥形瓶中,加入30 mL水,置于冰浴中冷却。待结晶完全后,过滤,晶体每次用2-3 mL50%冰冷过的乙醇洗涤2-3次。粗产物为白色晶体,经干燥后重约6-7 g,mp 92-93℃。粗产物可用25%的乙醇水溶液重结晶,mp 93℃。
2.香豆素-3-羧酸的合成
在100 mL圆底烧瓶中加入4 g香豆素-3-甲酸乙酯、3 g氢氧化钠、20 mL95%乙醇和10 mL水,加入几粒沸石,装上回流冷凝管,用水浴加热至酯溶解后,再继续回流15 min。稍冷后,在搅拌下将反应混合物加到盛有10 mL浓盐酸和50 mL水的烧杯中,立即有大量白色结晶析出。在冰浴中冷却使结晶完全。抽滤,用少量冰水洗涤晶体,压干,干燥后重约为3 g,mp 188℃。粗品可用水重结晶。
纯品香豆素-3-羧酸的熔点为190℃(dec)。
[思考题]
试写出利用Konvengel反应制备香豆素-3-羧酸的反应机理。反应中加入醋酸的目的是什么?
如何利用香豆素-3-羧酸制备香豆素?
« 课程考核
考核方式:考查
考核形式:试卷
成绩评定:百分制,平时成绩占70%,期末考试卷面成绩占30%。
« 教学材料
n 教材
u 《药物合成反应实验(全国高等医药院校药学类实验教材)》郭春主编 中国医药科技出版,20##-1-1,ISBN 9787506736145
n 主要参考资料
u 《基础有机化学》(第三版)(上、下册)邢其毅、徐瑞秋、周政编,高等教育出版社,2010,ISBN 9787040166378
u 《药物合成反应实验》刘玮炜编,化学工业出版社20##-9-1 ISBN 9787122145888
撰写人:孙晓华 审核人:
第二篇:药物合成实验的心得体会
学而时习之
学院:树达学院 学号:200921180113 姓名:李小凤
经过这几个实验,经过老师的教导,我学到了很多以前没学到的知识。才发现以前的操作有很多是不规范的,做实验也经常不会去动脑的。在几位老师的引导下,慢慢的也严格的要求自己的每一个动作的规范性,每一步骤的合理性,也学会分析在做实验时出现的问题,而不再仅仅是草草的动手做做实验,也会想想有什么方法可能更好,在哪个时间段用什么试剂,为什么要用到这个试剂,不用会出现什么现象等等……同时也对以前操作过的一些仪器或步骤的进行了回顾和温习,让自己更加熟悉每一个步骤,每一个细节。下面我将谈谈自己在三个实验中遇到的一些问题和学到了一点点知识。
实验一
甲基2,3,4,6-四-o-苯甲酰基-α-D-吡喃甘露糖苷的合成及结构表征
首先这个实验我们采用了薄层分析跟踪有机反应进程、柱层析分离纯化目标产物、了解到红外光谱法和核磁共振法。
1、薄层分析跟踪有机反应进程
在这个实验中我进一步的了解到薄层分析(TLC),即把样品点到薄层上,用适当的溶剂展开,根据各组分对固定相吸附能力的差异,从而使样品的各组分达到分离的目的。这个实验我们用的固定相是硅胶板,展开剂是乙酸乙酯和甲醇(5:3)混合溶液,然后在紫外灯光下描出点,显色方法是用的硫酸碳化法,并且在电炉上烘烤出点。利用TLC来检测反应的完全程度。Rf=原点中心至斑点中心的距离/原点中心至展开剂前沿的距离 ,用Rf来表示各个组分的位置。反应完全意味着我们的溶液里面没有了原料,即TLC的原料液点不会随展开剂展开,烤板后只有两个点反应点没有原料点才表示反应完全了。
TLC还用于检测纯目标产物,经过柱层析后,将流出物点到硅胶板上,和标准物对比,在紫外光下观察是否有荧光点,如没有则代表产物还没被层析出来,在给定的紫外线范围内只有产物才有荧光点,我们大概在14管的时候才出现淡淡的荧光点的,该点和标准物在统一展开线上,总层析了20管,从第14管到16管都有荧光点出现,后面的就没有什么了。即从14-16管中是纯产物。在观察荧光点时,还有苯甲酸的影响,它也有荧光点,但是经过烤板,苯甲酸的荧光点不会显示出来,所以不会对实验有影响。最后拷出的板上的点和标准物的点在统一直线上才算纯产物。
2、柱层析分离纯化目标产物
首先我们采用的是湿装,采用的洗脱剂是乙酸乙酯和石油醚(1:5),前期的装柱工作和我们平时的不同,这次多了一个溶剂球,溶剂求主要是用来缓冲洗脱剂的,防止添加洗脱剂时将装好的硅胶冲散。然后用滴管加入欲分离的物质,一定得沿壁加,不然加入的溶液不在同一平面,导致有些产物可能先出,分离的产物就不纯了。
3、红外光谱法和核磁共振法
红外:在3400左右有一个羟基峰,比较宽。除此之外,我们还做了核磁,有H谱、C谱、C-H相关谱图、H-H相关谱图.学会了根据H去找对应和相连的C或者H,然后确定化合物的结构,其中也不乏有些问题值得深思:
1、甲基α-D-吡喃甘露糖苷H谱中,为什么H被D取代后信号缺失?
答:因为当H被D取代后,原来有4个羟基变成3个羟基,少了一个羟基,所以信号缺失。
2、甲基2,3,4,6-四-o-苯甲酰基-α-D-吡喃甘露糖苷图中,在0处的峰是什么?
答:空气峰
3、甲基2,3,4,6-四-o-苯甲酰基-α-D-吡喃甘露糖苷图中,1.64处的信号是什么?
答:是氚代氯仿中的水
4、甲基2,3,4,6-四-o-苯甲酰基-α-D-吡喃甘露糖苷图中,为什么水的极性高达7.26,低到1.64?
答:因为水中含有氚代氯仿
5、为什么该实验用氚代氯仿而不直接用水?
答:假设用水,做出的图会看不到样品峰,而且氯仿只有一个H,可以溶解样品。
这个实验中我们还接触到新的仪器核磁和旋转蒸发仪,通过老师的指导下,我们学会了如何使用旋转蒸发仪浓缩和蒸发溶液,也体会到实验的每一步骤都不能随意的更改的,不然得到的产物可能不是我们所需要的构型或者得到的产物颜色不正常,所以加入原料的顺序不能随便更改。
实验二
对乙酰氨基酚的合成
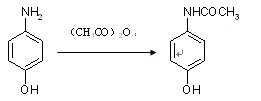
在这个实验中,我深刻的记得加人原料的顺序,错一步都会导致结果不同,例如加水,先加入水可以稀释醋酸酐,防止羟基也被氧化,使氨基的选择性强一些,若是后加水,则得到的产物的颜色比较深,产物中副产物较多,因为许多羟基可能也被氧化了。用旋转蒸发仪干燥,要干燥到物质不沾壁才算干燥完全。得到白色颗粒状结晶,然后我们用熔点仪测定了它的熔点180摄氏度。
思考题:
一、比较醋酐和醋酸作为酰化剂的差异?
答:1、醋酸酐容易断键,反应较快
2、醋酸酐能吸水,有利于反应的进行
3、无副反应,生成物较纯
酰化反应中加水30ml,有水存在,醋酐可选择性地酰化氨基而不与羟基反应,若以醋酸代替,则难以控制,反应时间长且产品质量差;产物转化率高,醋酐的转化率都在90%以上,醋酸一般不到60%反应条件温和,醋酐只需保证体系无水,室温即可,而醋酸需要浓硫酸脱水催化高温下才可反应系依旧无水,而醋酸反应后生成的水使得产物分离成为一个比较麻烦的问题,使用的分离手段复杂且会造成产物分解
产物纯净,由于醋酸的反应必须用浓硫酸催化,可能出现脱水多聚等副反应,醋酐则没有这个问题
二、亚硫酸氢钠在合成反应中的作用?
答:亚硫酸氢钠作为抗氧化剂,酚羟基易氧化,氧化物通常为黄色至红色的物质,
防止羟基被氧化,使获得的产物比较纯白,副产物少。
三、对乙酰氨基酚中的特殊杂质是如何产生的?
答:对乙酰氨基酚的特殊杂质:对氨基酚以及有关物质(对氨基酚、对氯乙酰苯胺、偶氮酚、苯醌)。
对氨基酚是对乙酰胺基酚合成中乙酰化反应不完全而引入的,也可能是因贮存不当使产品部分水解而产生的,是对乙酰氨基酚中的特殊杂质。对乙酰氨基酚在生产过程可能引入中间产物对氨基酚杂质;其次,本品在贮存过程中,受外部条件因素的影响,水解生成对氨基酚杂质。
实验三
(±)-?-苯乙胺的拆分
1、拆分原理:
用化学方法拆分外消旋体,其原理是用旋光性试剂把外消旋的对映异构体变成可分离的非对映异构体混合物,再利用非对映异构体的物理性质不同,将其分离。常用的方法是利用有旋光性的有机酸(或有机碱)与外消旋的有机碱(或有机酸)反应得到两种非对映异构体的盐的混合物,再利用它们在某种溶剂中的溶解度不同,用分步结晶法将它们分离。
2、(±)-?-苯乙胺的拆分
本实验采用L-(+)-酒石酸与(±)-?-苯乙胺反应,产生两个非对映异构体的盐的混合物,这两个盐在甲醇中的溶解度有显著差异,可以用分步结晶法将它们分离开来。S-(-)-?-苯乙胺-L-(+)-酒石酸盐
3、分别测定其熔点与旋光度
S- (-)-?-苯乙胺-L-(+)-酒石酸盐熔点:180-181 ºC(分解)。,测所得到的S-(-)-?-苯乙胺-L-(+)-酒石酸盐的旋光度为+0.138,文献值[a]D = -39.5?,计算其%ee=
实验的步骤很简单,可是当我们真正的用自己的双手去操作时,发现有很多现象是我们没想到,面临着一个个问题,老师耐心的为我们讲解,细心的给我们做演示,培养我们的动手和动脑能力,也鼓励我们多动脑多动手,不要畏惧的科学精神!同时老师严谨的态度和认真对待每一个细节的工作热情深深的感染着我们,也影响着以后我们的工作和学习。
在此我深深的感谢教导我的老师们,你们耐心的教导让我学到了许多知识。
我想说:老师,您们辛苦了!
药物合成实验
实验心得
 学院树达学院
学院树达学院
学号200921180113
姓名李小凤
-
3.药物的镇痛作用实验报告
实验题目药物的镇痛作用班级12级临七3班姓名廖梦宇学号20xx021320一实验原理扭体反应是药物镇痛作用实验的一个重要指标是指给…
- 药理学实验 镇痛药的镇痛作用
-
药理实验报告
药理学实验报告学校学院班级学号姓名指导老师元胡止痛片对小鼠镇痛抗炎镇痛活性研究组员指导老师摘要目的研究元胡止痛片是否具有镇痛抗炎的…
-
(甲组)镇痛药与解热镇痛药镇痛作用比较实验报告
镇痛药与解热镇痛药镇痛作用比较一实验目的1观察镇痛药和解热镇痛药的镇痛作用2学习药物镇痛作用的筛选方法扭体法二实验原理由于腹膜有广…
-
药理学实验报告.doc
巢湖职业技术学院ChaoHuvocationalandTechnicalCollege药理学实验报告供药学专业使用班级姓名组别指导…
-
药业公司仓库保管员工作总结
回顾这一年来的工作,我在公司领导和科长及各位同事的支持与帮助下,严格要求自己,按照公司的要求,较好地完成了自己的本职工作。现将一年…
-
人力资源和社会保障工作总结
XXX人力资源和社会保障局20xx年1-10月工作总结20xx年x月-10月,XXX人力资源和社会保障局深入贯彻落实科学发展观,坚…
-
20xx年城市管理工作总结
20xx年**镇城市管理工作总结和20xx年工作思路在过去的20xx年度中,我镇在区委区政府的统一领导下,在区城管执法局的有力指导…
-
清明扫墓活动总结
青年志愿者工作部缕缕春风,绵绵细雨,说不及对革命烈士的无限深情,道不尽对烈士英魂的无尽哀思。为了缅怀革命先烈,追忆革命历史,弘扬爱…
-
感恩教育工作总结
在当今社会,孩子在父母的心目中成了“小皇帝”、“小公主”,溺爱有加。而孩子自己也养成了唯我独尊、自私冷漠的心理趋势,孩子们渐渐远离…