生物信息学期末考试答案
一、名词
Bioinformatics:生物信息学——是一门综合运用生物学、数学、物理学、信息科学以及计算机科学等诸多学科的理论方法,以互联网为媒介、数据库为载体、利用数学和计算机科学对生物学数据进行储存、检索和处理分析,并进一步挖掘和解读生物学数据。
Consensus sequence:共有序列——决定启动序列的转录活性大小。各种原核启动序列特定区域内(通常在转录起始点上游-10及-35区域)存在共有序列,是在两个或多个同源序列的每一个位置上多数出现的核苷酸或氨基酸组成的序列。
Data mining:数据挖掘——数据挖掘一般是指从大量的数据中自动搜索隐藏于其中的有着特殊关系性的信息的过程。数据挖掘通常是利用计算方法分析生物数据,即根据核酸序列预测蛋白质序列、结构、功能的算法等,实现对现有数据库中的数据进行发掘。
EST:(Expressed Sequence Tag)表达序列标签——是某个基因cDNA克隆测序所得的部分序列片段,长度大约为200~600bp。
Similarity:相似性——是直接的连续的数量关系,是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比例的高低。
Homology:同源性——是两个对象间的肯定或者否定的关系。如两个基因在进化上是否曾具有共同祖先。从足够的相似性能够判定二者之间的同源性。
Alignment:比对——从核酸以及氨基酸的层次去分析序列的相同点和不同点,以期能够推测它们的结构、功能以及进化上的联系。或是指为确定两个或多个序列之间的相似性以至于同源性,而将它们按照一定的规律排列。
BLOSUM:模块替换矩阵——是指在对蛋白质数据库搜索时,采用不同的相似性分数矩阵进行检索的相似性矩阵。以序列片段为基础,从蛋白质模块数据库BLOCKS中找出一组替换矩阵,用于解决序列的远距离相关。在构建矩阵过程中,通过设置最小相同残基数百分比将序列片段整合在一起,以避免由于同一个残基对被重复计数而引入的任何潜在的偏差。在每一片段中,计算出每个残基位置的平均贡献,使得整个片段可以有效地被看作为单一序列。通过设置不同的百分比,产生了不同矩阵。
PAM(Point Accepted Mutation):突变数据矩阵PAM即可接受点突变——指1个PAM表示100个残基中发生一个残基突变概率的进化距离。在序列比对中,能够反映一个氨基酸发生改变的概率与两个氨基酸随机出现的概率的比值的矩阵。 Contig:叠连群——是指一组相互两两头尾拼接的可装配成长片段的DNA序列克隆群,也指彼此间可通过重叠序列而连接成连续的、扩展的、不间断的DNA序列的交叠片段产物。通过比对不同的序列,我们能够发现片段的顺序,并且contigs能被添加、删除、重排列来形成新的序列。
Phylogenetic tree:系统发生树又称为演化树(evolutionary tree)——是表明被认为具有共同祖先的各物种间演化关系的树,是一种亲缘分支分类方法。在树中,每个节点代表其各分支的最近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间)。它用来表示系统发生研究的结果,用它描述物种之间的进化关系。
In Silico Cloning:电子克隆——是近年来发展起来的一门基于表达序列标签(ESTs)的快速克隆基因的新技术,其利用种子序列从EST及UniGene数据库中搜索相似性序列,进行拼装、检索、分析等,以此获得目标基因的全长cDNA,在此基础上也能够实现基因作图定位。
二、问题思考
1、生物信息学这门学科是如何发展起来的?
答:生物学数据爆炸式增长
生物大分子数据库相继建立
生物技术与计算机技术并行飞速发展
Internet的广泛应用
人类基因组计划(HGP)的推动
生物信息学的产生是生命科学发展的必然。
2、举例说明生物信息学的主要应用?
答: a. 获取各种生物的全基因组及其他数据;
b. 新基因发现;
c. 单核苷酸多态性分析;
d. 基因组中非编码区域的结构与功能;
e. 从基因组水平研究生物进化及其他遗传语言的可能;
f. 全基因组的比较研究;
g. 基因功能预测;
h. 遗传疾病的研究以及关键基因鉴定;
i. 蛋白质组学研究;
j. 新药设计和定向化酶;
k. 生物芯片.
3、为什么说生物信息学是大规模研究生命科学的利器?
答:生物信息学主要是一门研究生物学系统和生物学过程中信息流的综合系统学科,是综合运用生物学、数学、物理学、信息科学以及计算机科学等诸多学科的理论方法,以互联网为媒介、数据库为载体、利用数学和计算机科学对生物学数据进行储存、检索和处理分析,并进一步挖掘和解读生物学数据。目前,其核心是基因组信息学,包括基因组信息的获取、处理、存储、分配和解读。还包括:蛋白质空间结构模拟、预测和药物分子设计;软件开发和方法学研究。未来,生物信息学将进一步揭示生命系统的复杂性、遗传语言、基因表达谱、基因组、蛋白质组、代谢组、细胞信号组、系统生物学等等。因此,生物信息学是大规模研究生命科学的利器。
4、生物信息学涉及的生物大分子信息有哪些?
答:涉及的有:
1)核算序列DNA
包括:基因组序列、基因序列、cDNA、EST、碱基修饰、DNA功能模块/位点(如启动子、剪接体、表达调控位点等)。
2)蛋白质Protein
包括:氨基酸组成、氨基酸序列、理化性质、原子坐标、二级结构、模体、结构域、功能域/位点、3D结构。
5、在大分子序列分析中,为何局部比对比全局比对更有意义?
答:全局比对(global alignment)——指全长序列比对,用于相似性很高的序列间的分析。 局部比对(local alignment)——指生物分子序列常常是局部具有较高的相似性,呈板块分布。此法用于整体相似性较低的序列分析,灵敏度高。
原因:
1)全局比对是沿整个长度实现序列之间匹配的最大化,尝试对齐整个序列。而局部比对是对动态规划算法的修改,是给两个序列之间得分最高的地方进行匹配,集中在寻找相似度高的序列的延伸。因此相比而言,在序列分析中将未知序列同已知序列进行相似性比较,局部比对的准确性比全局比对更高。因为要实现整个序列长度的相似性匹配,比起局部匹配分析带来的误差更大;
2)另外,与局部序列比对算法相比,全序列比对算法会导致一些局部序列相似性较高而全序列相似性很小,因为全序列的平均效应而将两者的相似性漏检。一般对于2个未知关系的序列,使用局部序
列比对工具要比用全序列比对工具好。而对于一个较长的序列和一个较短的序列的比对,也应该使用局部序列比对工具。
3)再则全局比对的最高分是最后一个,而局部比对的任何一个地方都可能是最高分,即任何地方都可以是对位起始点,可见局部比对操作更为灵敏。
4)应用范围上,全局比对仅适用于相似性很高的序列间分析,而局部比对一般用于相似性较低的序列分析,但是也可以用于高相似性序列分析,这样的分析结果会更加精准。
所以局部比对比全局比对更加有意义。
6、在大分子序列分析中,为何蛋白质的取代矩阵比核酸的取代矩阵更复杂?
答:取代矩阵(substitution matrix)的规则是“奖励匹配位点,罚扣不匹配位点”,故又称为计分矩阵(scoring matrix)。核算序列分析利用碱基取代矩阵,通过相似性比对匹配与否进行打分,便可以分析出其大致的碱基组成,特异位点等。而蛋白质序列利用其氨基酸残基取代矩阵分析,由于蛋白质的序列组成复制,而且蛋白质的功能是通过其三维高级结构来执行的,该结构又不一定处于静态,在行使功能的过程中,一般会发生相应的改变,所以氨基酸残基的进化取代不能简单地表述各种残基在结构和功能上的关系,所以要对蛋白质序列进一步的分析就需要更加复杂的取代矩阵。
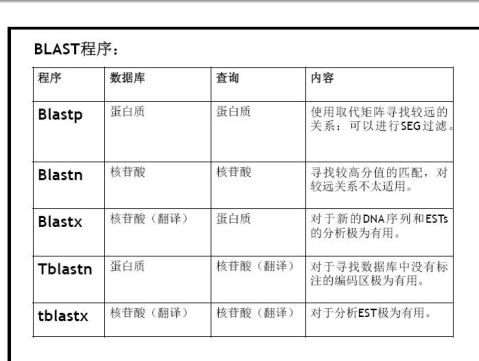
7、多重比对的用途?BLAST的用途?
答:多重比对的用途主要用于:
1) 系统演化分析,解释物种之间的进化关系;
2) 基因预测;
3) 蛋白质结构域的三级结构与二级结构,甚至是个别的氨基酸或核苷酸;
4) 研究一个家族中的相关蛋白质序列中的保守区域,进而分析蛋白质的结构和功能。
BLAST是现在应用最广泛的序列相似性搜索工具,主要用于:
1) 新DNA序列的发现、定位与分析、结构和功能预测;
2) ESTs的分析;
3) 寻找分析远源关系的蛋白质序列;
4) 实验设计如PCR Primer,Mutagenesis Studies,构建Profile(--谱)等;
5) 揭示相似性和同源性,发现系统发育的信息;
6) 寻找数据库中没有标注的编码区、发现保守区域、特定序列框等重要信息。
8、聚类分析的策略?
答:聚类分析(cluster analysis)是一组将研究对象分为相对同质的群组(clusters)的统计分析技术。其策略方法为:
先将多个序列两两比对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树,对关系密切的序列进行加权;然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止。
第一步:点击File→Load Sequences输入序列文件。
第二步:点击Alignment设定比对的一些参数。
第三步:点击Alignment→Do Complete Alignment开始序列比对。
第四步:点击File→Save Sequence as...比对完成,选择保存结果文件的格式。
9、电子克隆比传统的实验克隆有何优势?为何能实现电子克隆?
答:电子克隆利用种子序列从EST及UniGene数据库中搜索相似性序列,进行拼装、检索、分析等,以此获得目标基因的全长cDNA,在此基础上也能够实现基因作图定位。
其相比实验克隆所具有的优势有:
1) 实验进程短、快捷、设备简单;
2) 成本低、得率高、针对性强等;
3) 对操作人员技术要求不高;
4) 另外运用电子克隆的方法延伸得到的cDNA几乎囊括了所有疑似为目的基因的cDNA序列。 能实现电子克隆是因为:EST数据库的不断完善,使得电子克隆策略已成为克隆新基因的重要方法。 从GenBank的核酸(nr)数据库中检索已测序列生物的目的基因,获得目的基因cDNA序列,以该序列为模板对另一种未测序列生物EST数据库进行BLAST检索,获得与之部分同源的EST群,从中选取一条EST作为种子序列BLAST检索该生物的EST数据库,将检出与种子序列同源性较高或有部分重叠的EST序列拼接组装为重叠群(contig),再以此重叠群序列重复以上BLAST检索过程,反复进行EST重叠群序列的拼接和比对,直至检出所有的重叠EST或重叠群不能继续延伸,最终获得未测序列生物基因的cDNA全序列。
10、蛋白质分子结构的层次?相应的分析工具?
答:蛋白质一级结构分析:
1) ProtParam:蛋白质理化参数检索;
2) ProtScale:蛋白质亲疏水性分析;
3) coiled-coil 卷曲螺旋预测。
蛋白质二级结构预测:二级结构指α‐helix,β‐sheet,无规则卷曲(coil),motif等组件。 预测方法:
1) 神经网络、遗传算法、机器学习等;
2) 与已知二级模板建立序列谱矩阵(profile matrix)、PSI‐BLASTP;
3) 与同源蛋白多重比对。
模式和序列谱分析:EBI:InterProScan
整合出的部分数据库有:
Proside 蛋白质结构域、家族和功能位点;
Pfam 蛋白质家族比对;
TMHMM 跨膜区预测。
蛋白质三级结构预测:
实验测定方法:X-ray、NMR、Cryo-EM;
理论预测方法:同源建模、折叠识别、从头计算。
三、综合分析
1、DNA序列的鉴定策略
答:鉴定三步骤:
1) 找到序列中的非编码区
编码区与非编码区显著不同,重复序列和低复杂序列排除基因的可能性,首先屏蔽掉。屏蔽重复序列的分析程序有:RepeatMasker, XBLAST, CENSOR等。此外,确定待检序列是否真实(载体污染,宿主序列污染,纯度因素等),载体序列污染分析程序有:NCBI / VecScreen;EMBL / Blast2 EVEC。
2) 找基因
根据基因特征信号,如保守序列(启动子,CpG岛)、起始和终止密码子、polyA,碱基频率,密码子偏好,EST。原核生物采用可读框ORF检测基因非常有效。
CpG岛的预测工具:EMBL-EBIK的在线工具CpGPlot;
转录终止信号的预测方式:真核生物基因末端有终止子信号,在mRNA终止密码子下游具有polyA加
尾信号AATAAA,可用于基因终止位点的预测。在线预测工具:POLYAH;
启动子预测分析工具:TRES、Neural network、Dragon promoter finder、PromoterScan;
可读框ORF=起始密码子ATG——终止密码子TGA或TAG或TAA。
开放读框的识别分析程序有:ORF Finder (NCBI), GenScan, GenomeScan。
采用mRNA序列预测基因:以公共数据库获得mRNA /cDNA,从基因组序列预测基因,在线预测工具(NCBI) Spidey。
3) 鉴定找到的基因
建立基因模型以便核对,同源性搜索增加可信度
2、蛋白质结构分析和预测的策略
答:策略为:
1) 在数据库中搜寻与蛋白质序列相似的模板;
2) 查询序列和已知三维结构的蛋白质序列的相似性比对;
3) 如果符合相似则直接进行结构比较建模;
4) 如果不相似则先进行蛋白质家族、功能域、聚类分析,再与已知的蛋白质结构比对,有关系的才进行比较建模;
5) 若还是不相关,则对蛋白质序列进行结构分析,对可以预想出其结构的蛋白质预测其三维结构,对无法预想出结构的蛋白质在实验室中进行进一步结构分析。
知识点
生物信息学研究的基本方法
?生物学数据库的建立
?生物学数据的检索
?生物学数据的处理
?生物学数据的利用
生物信息数据的存储格式
一般由两/三部分组成:纪录信息、特性注释、序列本身
FASTA格式(序列最简单注释)
?序列文件的第一行是由大于符号(>)打头的任意文字说明,主要为标记序列用。
?从第二行开始是序列本身,标准核苷酸符号或氨基酸单字母符号。通常核苷酸符号
大小写均可,而氨基酸一般用大写字母。
?文件中和每一行都不要超过80个字符(通常60个字符)。
GenBank和EMBL数据库基本数据的格式
序列名称、长度、日期
序列说明、编号、版本号
物种来源、学名、分类学位置
相关文献作者、题目、刊物、日期
序列特征表
碱基组成
序列本身(每行60个碱基)
PDB格式
记录除了原子坐标外,还包括物种来源、化合物名称、结构递交以及有关文献等基本注释信息。此外,还给出分辨率、结构因子、温度系数、蛋白质主链数目、配体分子式、金属离子、二级结构信息、二硫键位置等和结构有关的数据。
蛋白质序列的格式
FASTA、序列文件格式、PDB数据格式
一次数据库
直接来源于实验获得的原始数据,只经过简单的归类、整理和注释。
一级核酸数据库:GenBank数据库、EMBL数据库、DDBJ数据库
一级蛋白质序列数据库:SWISS-PROT库、PIR库
一级蛋白质结构数据库:PDB数据库
二次数据库
在一级数据库、实验数据、文献数据和理论分析的基础上,针对不同的研究内容和需要,对生物学知识和信息的进一步整理得到的数据库。
人类基因组图谱库GDB、转录因子和结合位点库、TRANSFAC、蛋白质序列功能位点数据库Prosite等。 蛋白质数据库
序列数据库(序列及其注释):
SWISS-PROT、PIR (protein information resource)、NCBI(其功能和应用范围快速拓展) 模体和结构域数据库(结构域、功能域):
PROSITE、Pfam (protein families database of alignments and HMMs)
结构数据库:
PDB (protein databank)
蛋白质分类数据库:
SCOP、CATH、FSSP
PDB是目前最主要的收集生物大分子(蛋白质、核酸和糖,以及病毒)三维结构的数据库,是通过X射线单晶衍射、核磁共振、电子衍射等实验手段确定的蛋白质、多糖、核酸、病毒等生物大分子的三维结构数据库。
NCBI数据库检索系统Entrez
Entrez是NCBI开发的基于WWW的数据库检索工具,它可以用来搜索20多个集成在NCBI中的数据库信息。
数据库搜索:BLAST & FASTA
多序列比对工具
Clustal W:对DNA和蛋白质进行多序列联配并且生成亲缘树的工具。
EMBL:提供在线的基于万维网界面的ClustalW服务:
对Clustal W的结果进行观察的程序为:njplotWIN95, treeview, 等
构建进化树------基于大分子序列进化
分子系统发育:DNA在进化过程中积累突变,从而导致不同株系后代的DNA、RNA和蛋白质的分支。这个原则被用于进化树的构建。
进化树构建的基本步骤
1、多序列比对(自动或手动):用Clustal,有些软件已整合上Clustal, 如MEGA。
2、确定建树方法(取代模型):距离(UPGMA, NJ, ME)、最大节约(MP)、最大似然(ML),
3、建树;
4、进化树评估。
电子克隆
7.1 利用UniGene数据库进行序列电子延伸
7.2 从数据库中获取cDNA全长序列
7.3 序列拼接
本地拼接软件
Windows:Sequencher, DNAstar, ?
Unix: CAP3, Phrap, TIGR Assembler, Velvet, ?
在线服务:CAP3 网址
7.4 基因的电子表达谱分析
7.5 核酸序列的电子基因定位分析
蛋白质序列的获取
直接测序:Edman,蛋白质组技术
翻译编码的DAN序列:ORF,EBI protein machine搜索或检索数据库
同源建模是将目标序列在蛋白质结构数据库(PDB)比对搜索, 找出最好的
模板来构建新的结构, 再做能量最小化运算, 获得接近”真实”的蛋白质结构.
ExPASY提供三种生物信息学蛋白结构预测工具
1 Homology modeling;同源建模(25%以上一致性被认为有相似的结构)
2 Threading; 串线法(一致性低于30%时)
3 ab initio从头算(基于能量最低原则,分子力学、分子动力学)
同源建模的基本步骤
1 同源的参考蛋白搜索(PDB)
2 确定结构保守区: 如果目标蛋白有2个以上已知结构的参考蛋白,可将之叠加确定保守区,若仅一个有空间结构则做多重比对.
3 蛋白主链建模: 保守区主链坐标直接来自参考蛋白的, 环区可用片段搜索或自动生成.
4 侧链安装: 在转子文库中挑选最佳残基侧链构象组合.
5 优化处理: 根据分子动力学和分子力学.(能量最小化计算)
6 合理性检测: 常用Profiles-3D检测.
PubMed文献检索
PubMed是美国国家医学图书馆下属的国家生物技术信息中心(NCBI)开发的、基于WWW的查询系统:
1.如何理解生物信息语言的复杂性和生物信息学的局限性?
答:物体或者事物的属性,分为单一或者极度复杂,他们可通过任何方式,比如声音、光波、电波、颜色、行为、温度、气体、形态、能量等,传递到与之关联的事物的外界,却又得到多种应答:沟通、接纳、排斥、刺激。
2、几种常用的序列格式:
①GenBank序列格式 ②GCC序列格式 ③EMBL序列格式 ④ASN.1序列格式
⑤PIR/CODATA序列格式 ⑥SwissProt序列格式 ⑦Plain/ASCII.Staden序列格式
⑧FASTA序列格式 ⑨NBRF序列格式 ⑩GDE格式
⑾Intelligenetics序列格式 ⑿PDB格式
-
中学生期末评语)
学期期末评语2记得李嘉成说过其实每个人都可以有高远的梦想区别只在于当你失败时能否从自怨自艾的心境中走出来当你成功时能否在此基础上继…
- 期末给学生的评语
-
20xx期末学生评语大全
20xx期末学生评语大全一引导学生培养坚强毅力的评语1在赛场上你奋力拼博是班级和学校的骄傲你总是用优异的体育成绩向老师和同学们报喜…
-
期末学生评语
期末学生评语期末学生评语拥有健康体魄的同时祝愿你拥有健全思想拥有短暂奋发的同时祝愿你拥有坚韧不拔拥有侠肝义胆的同时祝愿你拥有方式方…
-
20xx学生期末评语汇编
20xx学生期末评语汇编gt一年级学生期末评语汇编1看到你总感觉你像个小哈利波特聪明懂事惹人喜爱每次见到老师你总是热情地向老师打招…
-
生物信息学专业实习总结范文
《浙江大学优秀实习总结汇编》生物信息学岗位工作实习期总结转眼之间,两个月的实习期即将结束,回顾这两个月的实习工作,感触很深,收获颇…
-
生物信息学小结
1.什么是(基因)生物信息学?目前一般意义的生物信息学是基因层次的它是一个包含着基因组信息的获取、处理、存储、分配、分析和解释的所…
-
高级生物统计学学习心得
高级生物统计学课程学习总结摘要经过一学期对生物统计学的学习我对生物统计学有了进一步的理解本文主要讲述了本学期学习生物统计之后我对生…
-
生物信息学实习一
实习一序列查询实验目的1了解三大生物信息中心的资源2学会用Entre系统查找目标序列3学会用SRS系统查找目标序列实验内容一三大生…
-
我眼中的生物信息学
我眼中的生物信息学学院:外国语学院年级:10级班级:商务一班姓名:学号:一、生物信息学的概念从广义上来说,生物信息学从事对基因组研…