哺乳动物组织基因组DNA提取实验报告



二.实验报告



第二篇:重组质粒实验报告
重组质粒的构建、转化、筛选和鉴定
实验目的:
学习在实现DNA体外重组过程中,正确选择合适的载体和限制性内切酶并能对限制性核酸内切酶对载体和目的DNA进行切割,产生利于连接的合适末端。
学习设计构建重组DNA分子的基本方法,掌握载体和外源目的DNA酶切的操作。
学习利用T4DNA连接酶把酶切后的载体片段和外源目的DNA片段连接起来,构建体外DNA分子的技术,了解并掌握几种常用的连接方式。
掌握利用Cacl2 感受态细胞的方法。
学习掌握热击法转化E.coli的原理和方法。
掌握α互补筛选法和PCR检测法筛选重组子的方法。并鉴定体外导入目的DNA片段的大小。
学习和掌握PCR反应的基本原理和操作技术,了解引物设计的基本要求。
实验原理:
外源DNA与载体分子的连接即为DNA重组技术,这样重新组合的DNA分子叫做重组子。重组的DNA分子式在DNA连接酶的作用下,有Mg2+、ATP存在的连接缓冲系统中,将分别经限制性内切酶酶切的载体分子和外源DNA分子连接起来。将重组质粒导入感受态细胞中,将转化后的细胞在选择性培养基中培养,可以通过α互补筛选法筛选出重组子,并可通过酶切电泳及PCR检验的方法进行重组子的鉴定。
重组子的构建
酶切时首先要了解目的基因的酶切图谱,选用的限制性内切酶不能目的基因内部有专一的识别位点,否则当用一种或两种限制性内切酶切割外源工体DNA时不能得到完整的目的基因。其次要选择具有相应的单一酶切位点质粒或者噬菌体载体分子。常用的酶切方法有双酶切法和单酶切法两种。本实验采用单酶切法,即只用一种限制性内切酶切割目的DNA片段,酶切后的片段两端将产生相同的黏性末端或平末端,再选用同样的限制性内切酶处理载体。在构建重组子时,除了形成正常的重组子外,还可能出现目的DNA片段以相反方向插入载体分子中,或目的DNA串联后再插入载体分子中,甚至出现载体分子自连,重新环化的现象。单酶切法简单易行单是后期筛选工作比较复杂。各种限制性内切酶都有去最佳反应条件,最主要的因素是反应温度和缓冲液的组成,在双酶切体系中,限制性内切酶在使用时应遵循“先低盐后高盐,先低温后高温”的原则进行反应。
(要达到高效率的连接,必须酶切完全,酶切的DNA数量要适当。另外,酶切反应的规模也取决于需要酶切的DNA的量,以及相应的所需酶的量。可以适当增加酶的用量,但是最高不能超过反应总体积的10%,因为限制性核酸内切酶一般是保存在50%甘油的缓冲液中,如果酶切反应体系中甘油的含量超过5%,就会抑制酶的活性。)
连接反应总是紧跟酶切反应,外源DNA片段与载体分子连接的方法即DNA分子体外重组技术主要依赖限制性核算内切酶和DNA连接酶催化完成的。DNA连接酶催化两双链DNA片段相邻的5’-磷酸和3’-OH间形成磷酸二酯键。在分子克隆中最有用的DNA连接酶是来自T4噬菌体的T4 DNA连接酶,它可以连接黏性末端和平末端。连接反应时,载体DNA和外源DNA的摩尔数之比控制在1:(1~3)之间,可以有效地解决DNA多拷贝插入的现象。反应温度介于酶作用速率和末端结合速率之间,一般是16℃,用常用的连接时间为12-16h。
感受态细胞的制备及质粒转化
构建好的重组DNA转入感受态细胞中进行表达的现象就是转化。能进行转化的受体细胞必须是感受态细胞,即受体细胞最容易接受外源DNA片段实现转化的生理状态,它决定于受体菌的遗传特性,同时与菌龄、外界环境等因素有关。人工转化是通过人为诱导的方法使细胞具有摄取DNA的能力,或人为地将DNA导入细胞内,该过程与细菌自身的遗传控制无关,常用热击法,电穿孔法等。能否实现质粒DNA的转化还与受体细胞的遗传特性有关,所用的受体细胞一般是限制修饰系统的缺陷变异株,即不含限制性内切酶和甲基化酶的突变株。
目前常用的感受态细胞制备方法有CaCl2法,制备好的感受态细胞可以加入终浓度为15%的无菌甘油,-70℃可保存半年至一年。经过CaCl2处理的细胞细胞膜通透性增加,允许外源DNA分子进入。在低温下,将携带有外源DNA片段的载体与感受态细胞混合,经过热击或电穿孔技术,使载体分子进入细胞。进入受体细胞的外源DNA分子通过复制、表达,使受体细胞出现新的遗传性状。将这些转化后的细胞在选择性培养基上培养,即可筛选出重组子。本实验以E.coli DH 5α菌株为受体细胞,用CaCl2处理,使其处于感受态,然后将重组后的PUC19质粒在42℃下热击90s,实现转化。
重组子的筛选鉴定
重组DNA转化宿主细胞后,并非所有的受体细胞都能被导入重组DNA分子,一般仅有少数重组DNA分子能进入受体细胞,同时也只有极少数的受体细胞在吸纳重组DNA分子之后能良好增殖。并且它们是与其他大量未被转化的受体菌细胞混杂在一起。再者,在这些被转化的受体细胞中,除部分含有我们所期待的重组DNA分子外,另外一些还可能是由于载体自身或一个载体与多个外源DNA片段形成非期待重组DNA分子导入所致。因此必须使用各种筛选及鉴定手段区分转化子与非转化子,并从转化的细胞群体中分理出带有目的基因的重组子。本实验中采用的方法是平板筛选法电泳筛选法及PCR检测方法。
抗药性筛选主要用于重组质粒DNA分子的转化子的筛选,而不含重组质粒DNA分子的受体菌则不能存活,α互补筛选法是根据菌落颜色筛选含有充足质粒的转化子。质粒PUC19携带有氨苄青霉素抗性基因(Ampr),在含有氨苄青霉素平板上筛选转化子。没有导入质粒PUC19的受体细胞,在含有氨苄青霉素的平板上不生长。质粒PUC19进入E.coli DH 5α后,通过α-互补作用,形成完整的β-半乳糖苷酶。在麦康凯培养基平板上,转化子利用β-半乳糖苷酶分解培养基中的乳糖产生有机酸,pH降低,培养集中的指示剂变红,转化子的菌落变红。不含质粒的E.coli DH 5α,没有β-半乳糖苷酶活性,不能利用培养集中的乳糖产生有机酸,而是利用培养集中的有机碳源,不使培养基pH降低,在不含有氨苄青霉素的麦康凯培养基上形成白色菌落。重组后的载体DNA因为目的基因的插入位点在PUC19乳糖利用基因内部,不能形成α-互补作用,所以也不能利用培养集中的乳糖产生有机酸,在含有氨苄青霉素的麦康凯培养基上形成白色菌落。
挑选在氨苄青霉素培养基上生长的白色菌落,通过扩增培养。因为许多菌落存在假阳性情况,在氨苄青霉素培养基上的白色菌落可能是导入的重组载体DNA菌落,也可能是载体自连后发生突变的菌落,所以还要鉴定转化子中重组质粒DNA分子的大小,可将重组的载体DNA提取出来,进行后续的酶切、电泳检验。
也以提取的重组质粒为模板,利用现有的引物,进行那个PCR扩增,检测个噢偶见的质粒是否是所期望的重组质粒。
PCR技术
PCR(Polymerase chain reaction)即聚合酶链式反应,其原理类似于DNA的体内复制,又叫做“体外基因扩增”。反应体系包括:模板DNA、寡核苷酸引物、Mg2+,4种脱氧核糖核酸(dNTP)、DNA聚合酶和合适的缓冲体系。反应基本程序包括DNA变性、引物复性及引物延伸三个基本过程。这三个反应构成一个循环,反复进行。每一轮循环扩增的产物可作为下一轮扩增反应的模板。理论上每一轮循环可使DNA数量增加一倍,反复30次,特异DNA序列片段以指数方式可扩增105~106。通过此技术可以使非常微量的DNA产生大量的PCR产物,因此这个技术是一项在体外大量生产目的基因的技术。
现在普遍通用的是一种从水生嗜热杆菌中提取的Taq DNA聚合酶,而且大大提高了扩增片段的特异性、灵敏性和扩增效率。反应中用的引物有两段,一条称为上游引物或者正向引物,一条称为下游引物或者反向引物。引物的设计十分重要,在设计时应遵循以下原则:长度一般为18到24个碱基;G+C的百分含量在40%~60%;单条引物内部避免含有二级结构,避免形成引物二聚体;最好以1到2个G或C碱基开始或结束;引物的5’端可以被修饰,但是3’端绝对不能进行任何修饰,而且引物的3’端要避开密码子的第三位。模板的浓度不能太大,dNTP的浓度为2.5mmol/L,金属离子的浓度为1.5mmol/L,缓冲液为pH为7.2~8.0的Tris—HCl溶液。
实验材料:
菌株:E.coli DH 5α
培养基: LB培养基、麦康凯培养基(加入氨苄青霉素)
试剂材料:
酶切反应:(DNA PUC19 质粒,酶切10×buffer,Hind Ⅲ,重蒸水,λ DNA。)
连接反应:(酶切后的DNA PUC19 质粒和λ DNA,连接10×buffer,T4 连接酶,重蒸水。)
感受态细胞的制备:(0.1M CaCl2 )
转化:(连接液和感受态细胞,0.1M CaCl2,冰块。)
重组菌的挑选、检验:试剂盒(含有Rnase A的溶液Ⅰ,溶液Ⅱ,溶液Ⅲ,HB buffer,DNA washing buffer,elution buffer),酶切后的DNA PUC19 质粒,试剂盒抽提的DNA,酶切10×buffer,Hind Ⅲ,重蒸水,琼脂糖,TAE缓冲液。上样缓冲液,EB 染液。
PCR检测 : 10×PCR buffer,dNTP,模板,引物1,引物2,水,Taq DNA聚合酶,琼脂糖,TAE、溴化乙锭(EB)
4. 仪器器材:
20、200、1000ul 的枪和枪尖,1.5 ml的Ep管,恒温培养箱,水浴锅,平板,三角瓶,试管,接种环,牙签,酒精灯,火柴,冰箱,离心机,电泳槽,紫外仪,PCR扩增仪
试验流程
载体与外源片段的酶切→连接→感受态细胞的制备→质粒转化与重组子的筛选→重组子的挑取与复证→重组质粒的提取与电泳检测→PCR检测
注意事项:
本实验属于微量操作,用量极少的步骤必须严格注意吸取量的准确性并确保样品全部加入反应体系中。
实验中所用塑料器材都必须是新的,并且经过高温灭菌,操作时打开使用,操作过程中要注意环境干净,戴手套操作,尽量减少走动,缩短Ep管开盖的时间。
不论是酶切还是连接反应,加样的顺序应该是,先加重蒸水,其次是缓冲液和DNA,最后加酶。且前几步要把样品加到管底的侧壁上,加完后用力将其甩到管底,而酶液要在加入前从-20℃的冰箱取出,酶管放置冰上,取酶液时吸头应从表面吸取,防止由于插入过深而使吸头外壁沾染过多的酶液。取出的酶液应立即加入反应混合液的液面以下,并充分混匀。
Ep管的盖子应盖紧,防止水浴过程中水汽进入管内,并做好标记以防样品混淆。
转化过程要防止杂菌和其他DNA的污染,整个操作过程应在无菌条件下进行。
电泳时使用的缓冲液最好是现配现用,以免影响电泳效果。
制备凝胶时,应避免琼脂糖溶液在微波炉里加热时间过长,否则溶液将会暴沸蒸发,影响琼脂糖浓度。制胶时要除去气泡。拔梳子时要特别小心,以防凝胶与支持物脱离。
上样时要小心操作,避免损坏凝胶或将样品槽底部的凝胶刺穿。也不要快速挤出吸头内的样品,避免挤出的空气将样品冲出样品孔。
溴化乙锭是一种强烈的诱变剂,有毒性,使用含有这种染料的溶液时,应带上乳胶手套进行操作。勿将溶液滴洒在台面上,实验结束后用水彻底冲洗干净。
紫外线对眼睛和皮肤均有伤害,对眼睛尤甚。观察电泳条带时要确保紫外光源得到适当遮蔽,并应戴好目镜或眼罩,避免皮肤直接暴露在紫外线下。
实验中加样后应及时更换吸头,以避免试剂的污染。
PCR反应高度灵敏,应设法避免污染,如戴一次性手套操作,使用一次性PCR管和吸头,反应加样区应与DNA模板制备区及PCR产物电泳检测区分开。
PCR管单加样时,对于非常微量的样品一定将样品加在管壁上或者液体中,防止漏样。
实际操作时,为了防止少加样,可以保存每次用过的枪尖,通过数枪尖知道自己加到哪一步了。
实验结果与分析
1、酶切电泳图的结果及分析
↓
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18

17,18泳道分别是标准λDNA酶切和自己的λDNA酶切后的产物的条带。15泳道为marker,16泳道是未酶切的λDNA的条带。12,13,14泳道分别是pUC19和商品pUC19酶切。
1泳道为我们的样品,是经酶切过的DNA PUC19 质粒,从图中可以看出只有有一条主带,这条是被Hind III切开的线性DNA PUC19 质粒,与DNA marker比较查资料可知分子量为2322,且可根据亮度看出含量不是很高。但是可以判断出我们组的载体质粒PUC19酶切很彻底。其他组的条带有些下面还有一条很浅的带是超螺旋DNA PUC19质粒,即没有被切开的质粒,分子量为2027。
2 重组子筛选结果
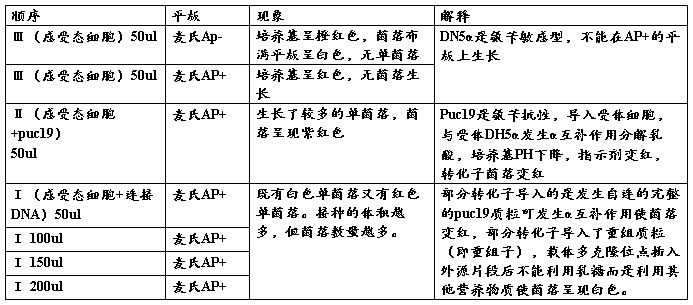
转化后,10个平板的菌落状况:
3 重组质粒电泳结果 ↓ ↓
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15


Re-pUC19电泳检测:(L1-2:lamda/HIII,L3-14:1-6组样品各2个;L15:空载体)
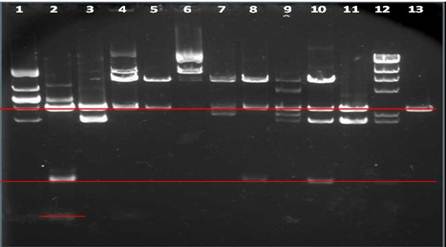
13,14泳道为本组重组质粒电泳结果。根据每条带的亮度可以看出DNA片段的含量。与DNA marker 对比带的位置可以比较DNA片段的大小。
与15泳道的空载体对照可以看出,我们筛选到的重组质粒中插入了外源DNA片段。重组质粒中分别插入了较小的基因片段(14)和较大的基因片段(13),对照DNA marker lamda/HIII,我们推测载体pUC19中插入的片段可能分别是125bp和6557bp大小的片段。结果还有待于进一步酶切验证和PCR检测。
4酶切验证电泳结果
下图分别是经过试剂盒提取的插入大片段和小片段的重组质粒的酶切验证电泳图。
↓
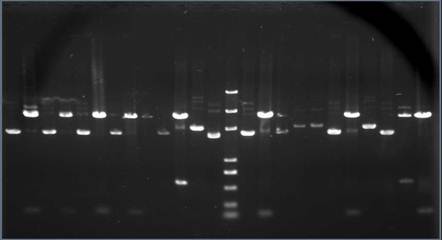
Re-pUC19的酶切鉴定大片段(1-11:各组样品;L12:lamda/HIII;L13:pUC19/III)
↓ ↓
 1 2 3 4 5 6 7 8 9 1011 121314
1 2 3 4 5 6 7 8 9 1011 121314
Re-pUC19的酶切鉴定小片段(1-12:各组样品;13:pUC19;14:DL2000 plus;15-25:各组样品)(DL2000 plus的大小分别为:5000,3000,2000,1000,750,500,250,100)
第一张图中4泳道是我们组插入大片段的重组质粒酶切电泳条带,第二张图中的7,8泳道分别是我们的插入小片段的重组质粒的酶切前后的电泳条带,其中酶切前的电泳结果作为对照。
从图中可以看出插入大片段的重组质粒酶切后( 4号泳道),上面有紧邻的两条带,与DNA marker lamda/HIII比较可知其分子量大小在6557bp左右,与上一步实验中的推测相符合。因此可以判定此重组质粒中插入了6557bp大小的DNA片段。理论上上方应该只出现一条带,而我们的外源DNA条带的上方紧邻着一条带,我们推测这是由于酶切体系中加入的酶的量太少而导致酶切不彻底的原因,使得样品中含有未酶切的重组质粒。下面的一条带是酶切后的线性PUC19质粒。
插入小片段的重组质粒酶切后产生了两条带(8号),与酶切前(7号)对照可知,上面一条带是酶切后的线性PUC19质粒,下面一条是插入的外源片段,与marker 对照,分子量大约为125,验证了上一步实验的推测,即插入了大小为125bp 的小片段外源DNA分子。从亮度上看,含量不是很高。
PCR电泳结果 ↓ ↓
1 2 3 4 5 6 7 8 9 10 11 12 18

(1-12 各组样品,18 DL2000 plus)
如图,11,12 号分别是为我们的564bp 和125bp 大小的片段的PCR产物电泳结果。以564bp片段为模板的扩增产物大小应为564+150=714,以125bp片段为模板的扩增产物大小应为125+150=275bp。与DNA marker 对比,可知我们的PCR产物大小分别为700bp左右和400bp左右。125bp外源片段的PCR产物大于125bp,而564bp的片段PCR产物电泳结果与理论值相同。我们推测产生此差异的原因是:增加了连接时候外源片段与载体的比例,出现了多个片段连接的情况。酶切时产生的125bp条带其实是两个125bp片段串联后又连接进载体的,所以以其为模板进行PCR时产物的大小应该是125+125+150=400bp而大于预期的125+150的275bp。564bp的外源片段是老师提供的标准片段,PCR结果与预期结果相同。PCR检测结果与酶切验证结果相同,因此实验结果可靠。
-
实验动物学实验报告
实验动物学实验报告一实验动物小鼠二操作流程抓取固定编号给药取血麻醉绝育解剖三具体操作1抓取抓取小鼠时右手抓住小鼠尾巴不要过于用力以…
-
实验动物学实验报告
实验一大小鼠的基本实验操作一实验目的通过实际操作掌握大小鼠的一般操作方法包括大小鼠的抓取和固定性别鉴定给药采血二实验动物昆明小鼠4…
-
实验动物学报告1
医学实验动物学实验报告(一)实验名称:小鼠的一般操作实验日期:姓名:学号:专业:一、实验目的和要求掌握小鼠实验的一般操作:动物的抓…
-
动物学实验报告
宁波大学考核答题纸20102011学年第1学期课号147L01D03课程名称医学动物实验学改卷教师李萍学号096080002姓名李…
-
实验动物学报告2
医学实验动物学实验报告二实验名称大鼠的一般操作实验日期姓名学号专业一实验目的和要求掌握大鼠的一般操作方法抓取和保定性别鉴别给药采血…
-
DNA提取实验报告
大肠杆菌和植物基因组DNA提取生科基彭健鹏20xx1412420xx1大肠杆菌DNA提取方案设计实验目的学习并掌握细菌基因组的提取…
-
浙大生化实验报告DNA的提取
实验报告课程名称生化实验甲指导老师成绩实验名称植物基因组DNA的提取实验类型生化定性实验同组学生姓名及纯度与含量的测定一实验目的和…
-
DNA提取实验报告
生物学大实验二实验名称质粒扩增提取和鉴定实验实验目的通过对智力扩增提取和鉴定试验的实验原理介绍和实际操作加深对分子生物学基本技术的…
-
动物肝脏中DNA的提取和鉴定
动物肝脏中DNA的提取和鉴定一实验目的1学习并掌握动物组织总DNA的提取方法及其原理2掌握琼脂糖凝胶电泳分离核酸的原理和方法3学习…
-
实验6 动物肝脏中DNA的提取
实验六动物肝脏中DNA的提取及定量测定一实验目的1学习和掌握用浓盐法从动物组织中提取DNA的原理与技术2了解常见生化组分提取技术3…
-
动物细胞融合 实验报告
姓名董朔晖系年级生命科学院20xx级学号20xx00230164科目细胞生物学实验实验题目动物细胞融合日期20xx0301实验二动…