天津大学物理化学知识点归纳
第一章 气体的pvT关系
一、 理想气体状态方程
pV=(m/M)RT= nRT (1.1)
或pVm=p(V/n)=RT (1.2)
式中p、V、T及n的单位分别为Pa、m3、K及mol。Vm=V/n称为气体的摩尔体积,其单位为m3·mol。R=8.314510J·mol-1·K-1称为摩尔气体常数。
此式适用于理想,近似于地适用于低压下的真实气体。
二、理想气体混合物
1.理想气体混合物的状态方程 (1.3)
pV=nRT=(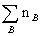 )RT
)RT
pV=mRT/Mmix (1.4)
式中Mmix为混合物的摩尔质量,其可表示为
Mmix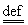
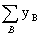 MB (1.5) Mmix=m/n=
MB (1.5) Mmix=m/n=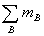 / (1.6)
/ (1.6)
式中MB为混合物中某一种组分B的摩尔质量。以上两式既适用于各种混合气体,也适用于液态或固态等均匀相混合系统平均摩尔质量的计算。
2.道尔顿定律
pB=nBRT/V=yBp (1.7)
P=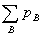 (1.8)
(1.8)
理想气体混合物中某一种组分B的分压等于该组分单独存在于混合气体的温度T及总体积V的条件下所具有的压力。而混合气体的总压即等于各组分单独存在于混合气体的温度、体积条件下产生压力的总和。以上两式适用于理想气体混合系统,也近似适用于低压混合系统。
3.阿马加定律
VB*=nBRT/p=yBV (1.9)
V=∑VB* (1.10)
VB*表示理想气体混合物中物质B的分体积,等于纯气体B在混合物的温度及总压条件下所占有的体积。理想气体混合物的体积具有加和性,在相同温度、压力下,混合后的总体积等于混合前各组分的体积之和。以上两式适用于理想气体混合系统,也近似适用于低压混合系统。
三、临界参数
每种液体都存在有一个特殊的温度,在该温度以上,无论加多大压力,都不可能使气体液化,我们把这个温度称为临界温度,以Tc或tc表示。我们将临界温度Tc时的饱和蒸气压称为临界压力,以pc表示。在临界温度和临界压力下,物质的摩尔体积称为临界摩尔体积,以Vm,c表示。临界温度、临界压力下的状态称为临界状态。
四、真实气体状态方程
1.范德华方程
(p+a/Vm2)(Vm-b)=RT (1.11)
或(p+an2/V2)(V-nb)=nRT (1.12)
上述两式中的a和b可视为仅与气体种类有关而与温度无关的常数,称为范德华常数。a的单位为Pa·m6·mol,b的单位是m3mol.-1。该方程适用于几个兆帕气压范围内实际气体p、V、T的计算。
2.维里方程
Z(p,T)=1+Bp+Cp+Dp+… (1.13)
 或Z(Vm, ,T)=1+B/Vm+C / Vm2 +D/ Vm3 +… (1.14)
或Z(Vm, ,T)=1+B/Vm+C / Vm2 +D/ Vm3 +… (1.14)
上述两式中的Z均为实际气体的压缩因子。比例常数B’,C’,D’…的单位分别为Pa-1,Pa-2,Pa-3…;比例常数B,C,D…的单位分别为摩尔体积单位[Vm]的一次方,二次方,三次方…。它们依次称为第二,第三,第四……维里系数。这两种大小不等,单位不同的维里系数不仅与气体种类有关,而且还是温度的函数。
该方程所能适用的最高压力一般只有一两个MPa,仍不能适用于高压范围。
五、对应状态原理及压缩因子
1.压缩因子的对应式
Z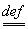 PV/(nRT) =pVm/(RT) (1.15)
PV/(nRT) =pVm/(RT) (1.15)
压缩因子Z是个量纲为1的纯数,理想气体的压缩因子恒为1。一定量实际气体的压缩因子不仅与气体的T,P有关,而且还与气体的性质有关。在任意温度下的任意实际气体,当压力趋于零时,压缩因子皆趋于1。此式适用于纯实际气体或实际气体混合系统在任意T,p下压缩因子的计算。
2.对应状态原理
Pr=p/pc (1.16)
Vr=Vm/Vm,c (1.17)
T=T/Tc (1.18)
pr、Vr、Tc分别称为对比压力、对比体积和对比温度,又统称为气体的对比参数,三个量的量纲均为1。各种不同的气体,只要有两个对比参数相同,则第三个对比参数必定(大致)相同,这就是对应状态原理。
第二章 热力学第一定律
一、 热力学基本概念
1. 状态函数
状态函数,是指状态所持有的、描述系统状态的宏观物理量,也称为状态性质或状态变量。系统有确定的状态,状态函数就有定值;系统始、终态确定后,状态函数的改变为定值;系统恢复原来状态,状态函数亦恢复到原值。
2. 热力学平衡态
在指定外界条件下,无论系统与环境是否完全隔离,系统各个相的宏观性质均不随时间发生变化,则称系统处于热力学平衡态。热力学平衡须同时满足平衡(△T=0)、力平衡(△p=0)、相平衡(△μ=0)和化学平衡(△G=0)4个条件。
二、热力学第一定律的数学表达式
1.△U=Q+W
或dU=ΔQ+δW=δQ-pambdV+δW`
规定系统吸热为正,放热为负。系统得功为正,对环境做功为负。式中pamb为环境的压力,W`为非体积功。上式适用于封闭系统的一切过程。
2.体积功的定义和计算
系统体积的变化而引起的系统和环境交换的功称为体积功。其定义式为:
δW=-pambdV
(1) 气体向真空膨胀时体积功所的计算
W=0
(2) 恒外压过程体积功
W=pamb(V1-V2)=-pamb△V
对于理想气体恒压变温过程
W=-p△V=-nR△T
(3) 可逆过程体积功
Wr=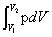
(4)理想气体恒温可逆过程体积功
Wr==-nRTln(V1/V2)=-nRTln(p1/p2)
(5)可逆相变体积功
W=-pdV
三、恒热容、恒压热,焓
1.焓的定义式
H U + p V
U + p V
2.焓变
(1)△H=△U+△(pV)
式中△(pV)为p V乘积的增量,只有在恒压下△(pV)=p(V2-V1)在数值上等于体积功。
(2)△H=
此式适用于理想气体单纯p VT变化的一切过程,或真实气体的恒压变温过程,或纯的液、固态物质压力变化不大的变温过程。
3. 内能变
(1)△U=Qv
式中Qv为恒热容。此式适用于封闭系统,W`=0、dV=0的过程。
△ U=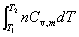 =
=
式中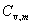 为摩尔定容热容。此式适用于n、CV,m恒定,理想气体单纯p、V、T变化的一切过程。
为摩尔定容热容。此式适用于n、CV,m恒定,理想气体单纯p、V、T变化的一切过程。
4. 热容
(1) 定义
当一系统由于加给一微小的热容量δQ而温度升高dT时,δQ/dT这个量即热容。
(2) 摩尔定容热容CV,m
CV,m=CV/n=( )V (封闭系统,恒容,W非=0)
)V (封闭系统,恒容,W非=0)
(3)摩尔定压热容Cp,m
Cp,m=
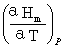 (封闭系统,恒压,W非=0)
(封闭系统,恒压,W非=0)
(4) Cp, m与 CV,m的关系
系统为理想气体,则有Cp, m—CV,m=R
系统为凝聚物质,则有Cp, m—CV,m≈0
(5)热容与温度的关系,通常可以表示成如下的经验式
Cp, m=a+bT+cT2
或Cp, m=a+b`T+c`T-2
式中a、b、c、b`及c`对指定气体皆为常数,使用这些公式时,要注意所适用的温度范围。
(6)平均摩尔定压热容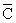 p,m
p,m
p,m=(T2-T1)
四、理想气体可逆绝热过程方程

 =1
=1
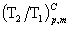
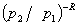 =1
=1
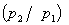
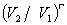 =1
=1
上式γ=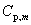 /,称为热容比(以前称为绝热指数),以上三式适用于为常数,理想气体可逆绝热过程,p,V,T的计算。
/,称为热容比(以前称为绝热指数),以上三式适用于为常数,理想气体可逆绝热过程,p,V,T的计算。
五、反应进度
ξ=△nB/vB
上式适用于反应开始时的反应进度为零的情况,△nB=nB-nB,0,nB,0为反应前B的物质的量。
νB为B的反应计算数,其量纲为1。ξ的单位为mol。
六、热效应的计算
1.不做非体积功的恒压过程
Qp=△H=
2.不做非体积功的恒容过程
Qv=△U=
3.化学反应恒压热效应与恒容热效应关系
Qp- Qv=(△n)RT
4.由标准摩尔生成焓求标准摩尔反应焓变
 =
=
5由标准摩尔燃烧焓求标准摩尔反应焓变
=—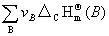
6. 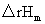 与温度的关系
与温度的关系
基希霍夫方程的积分形式
(T2)= (T1)+ 
基希霍夫方程的微分形式
d=△r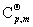 dT=
dT=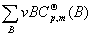
七、节流膨胀系数的定义式
μJ-T=(аT/аp)H
μJ-T又称为焦耳—汤姆逊系数
第三章 热力学第二定律
一、 卡诺循环
1. 热机效率
η=-W/Q1=(Q1+Q2)/Q1=(T1-T2)/T1
式中Q1和Q2分别为工质在循环过程中从高温热源T1吸收热量和向低温热源T2放出热量这两个过程的可逆热。此式适用于在两个不同的温度之间工作的热机所进行的一切可逆循环。
2.卡诺循环
所有工作于两个确定温度之间的热机,以可逆热机效率最大。
η1rηr
即是Q1/T1+Q2/T2 ≤0
式中T1、T2为高低温热源的温度。可逆时等于系统的温度。
二、热力学第二定律
1.克劳修斯说法
“不可能把热从低温物体传到高温物体而不产生其他影响。”
2.开尔文说法
“不可能从单一热源吸取热量使之完全转变为功而不产生其他影响。”
三、熵
1.熵的定义
d SδQ r/T
式中Q r为系统与环境交换的可逆热,T为可逆热δQ r时系统的温度。
2.克劳修斯不等式
dS
3.熵判据
△Siso=△Ssys+△Samb
式中iso、sys和amb分别代表隔离系统、系统和环境。在隔离系统中,不可逆过程即自发过程。可逆,即系统内部及系统与环境之间处于平衡态。在隔离系统中,一切自动进行的过程都是向熵增大的方向进行,这称为熵增原理。此式只适用于隔离系统。
四、熵变的计算
1.单纯的PVT变化
过程中无相变化和化学变化,W`=0,可逆。
△S= =
=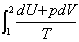 =
=
理想气体系统
△S=nCV,mln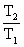 +nRln
+nRln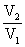 = nCp,mln- nRln
= nCp,mln- nRln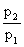 = n Cp ,m l n+ n CV ,m ln
= n Cp ,m l n+ n CV ,m ln
恒温(T1=T2)△S= nRln=- nRln
恒压(p1=p2)△S= nCp,mln= n Cp ,m l n
恒容(V1=V2)△S= nCV,mln= n CV ,m ln
凝聚相系统
△S=
恒容△S =
恒压△S=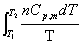
恒温△S=δQ r/T
2.相变化
可逆变化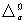 S=H/T
S=H/T
不可逆相变,通常设计一条要包括可逆相变步骤在内的可逆途径,此可逆途径的热温熵才是该不可逆过程的熵变。
3.环境熵差及隔离系统熵差的计算
△Samb= = Qamb/ Tamb=- Qsys / Tamb
= Qamb/ Tamb=- Qsys / Tamb
△Siso=△Samb+△Ssys
4.化学反应的标准反应熵
 =—
=—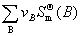
若在温度区间T1~T2内,所有反应物及产物均不发生相变化,则
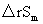 (T2)=(T1)+
(T2)=(T1)+
五、热力学第三定律
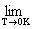
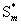 (完美晶体,T)=0
(完美晶体,T)=0
或 (完美晶体,0K)=0
上式中符号*代表纯物质。上述两式只适用于完美晶体。
六、亥姆霍兹函数
1. 亥姆霍兹函数定义式
AU-TS
式中A为系统的亥姆霍兹函数,U为系统的内能;TS为系统温度与规定熵的乘积。
2. 亥姆霍兹函数判据
dAT,V≤0
在恒温恒容且不涉及非体积功时,才能用△A判断过程的方向性。若△T,VA<0,则表明在指定的始终态(T,V相等)之间有自动进行的可能性;若△T,VA>0,则表明在指定的始末态之间处于平衡态。
3. 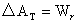
恒温可逆过程,系统的亥姆霍兹函数变化等于此过程的可逆功Wr。
七、吉布斯(Gibbs)函数
1.吉布斯(Gibbs)函数的定义式
GH-TS
H、A及G皆为组合函数,它们皆是系统具有广延性质的状态,而且皆具有能量的单位。状态一定,它们皆应有确定的数值,但它们的绝对值既无法测定,也无法求算。
2. 吉布斯(Gibbs)函数数据
dGT,P≤0
在恒温恒压且不涉及非体积功时,才可用△G来判断过程的方向性,在此条件下过程的方向性,在此条件下过程只能向吉布斯函数G减少的方向进行。
3. △GT,P=W`r
在恒温恒压下,过程的吉布斯函数等于始末状态间过程的可逆非体积功。在恒温恒压可逆的条件下,此等式才成立。
八、热力学基本方程
d U=T d S-p d V
d A=-S d T-p d V
d H=T d S-V d p
d G=-S d T+V d p
热力学基本公式的适用条件为封闭的热力学平衡系统的可逆方程。不仅适用于一定量的单相纯物质,或组成恒定的多组分系统发生单纯p、V、T变化的可逆过程,也可适用于相平衡或化学平衡的系统由一平衡状态变为另一平衡状态的可逆方程。
九、克拉佩龙方程
1.克拉佩龙方程
dp/dT=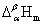 /(T
/(T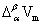 )
)
此方程适用于纯物质的α相和β相的两相平衡。
2.克劳修斯-克拉佩龙方程
dln(p/[p])=( △vapHm/RT2)dT
ln(p2/p1)=( △vapHm /R)(1/T1=1/T2)
此式适用于气-液(或气-固)两相平衡;气体可视为理想气体; (l)与(g)相比较可忽略不计;在T1~T2的温度范围内摩尔蒸发焓可视为常数。
(l)与(g)相比较可忽略不计;在T1~T2的温度范围内摩尔蒸发焓可视为常数。
对于气-固平衡,上式的△vapHm则应改为固体的摩尔升华焓。
十、吉布斯-亥姆霍兹方程
 =-U/T2
=-U/T2
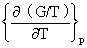 =-H/T2
=-H/T2
这两个方程分别表示了A/T在恒容下随T的变化及G/T在恒压下随T的变化。
十一、麦克斯韦关系式
-( /
/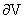 )S=(
)S=(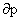 /
/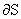 )V
)V
(/)S=(/)p
- (/)p=(/)T
(/)V=(/)T
这4个偏微分方程称为麦克斯韦关系式。物质的量恒定的单相纯物质,只有pVT变化的一切过程,上述4式皆可适用。对于混合系统,则要求系统的组成恒定,式中V及S分别为系统总体积及总的规定熵。
第四章 多组分系统热力学
一、 偏摩尔量
XB

其中X为广度量,如V,U,S
全微分式
dX=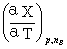 dT+
dT+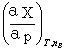 dp+
dp+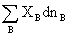
总和X=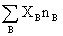
2.吉布斯—杜亥姆方程
在T、p一定条件下,=0
或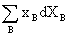 =0
=0
此处,xB指B的摩尔分数,XB指B的偏摩尔量。
3.偏摩尔量间的关系
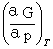 =V=
=V= =VB
=VB
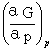 =-S
=-S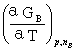 =-SB
=-SB
二、化学式
1、定义式
混合物(或溶液)中组分B的偏摩尔吉布斯函数GB又称B的化学势。
μBGB=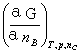
由热力学的4个基本方程可以得:
μB=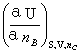 =
= =
=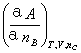 =
=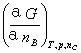
2.化学势判据

 ≤0
≤0 (dT=0,dV=0,Δw`=0)
(dT=0,dV=0,Δw`=0)
≤0(dT=0,dp=0,Δw`=0)
其中,μB(α)指α相内的B物质。
三、气体组分的化学势
1. 理想气体化学势
(1)纯理想气体的化学势为
μ*(pg)=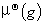 +RTln(p/
+RTln(p/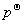 )
)
μ*(pg)表示纯理想气体在温度T、压力p时的化学势。是纯理想气体在标准压力=100kPa下的化学势,即标准化学势。
(2)混合理想气体中任一组分B的化学势为
μB(pg)= +RTln
+RTln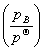
其中,pB=yB为B的分压。
2.真实气体化学势
(1)纯真实气体的化学势为
μ*(g)=+RTln +
+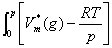 dp
dp
其中,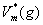 为该温度下纯真实气体的摩尔体积。低压下,真实气体近似认为是理想气体,故积分项为0。
为该温度下纯真实气体的摩尔体积。低压下,真实气体近似认为是理想气体,故积分项为0。
(3) 真实气体混合物中任一组分B的化学势为
μB(g)=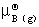 +RTln
+RTln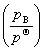 +
+ dp
dp
其中,VB(g)为真实气体混合物中组分B温度T及总压p总 下的偏摩尔体积。
四、拉乌尔定律与亨利定律
1.拉乌尔定律
pA= xA
xA
式中pA为溶液A的蒸汽压;为纯溶剂在同样温度下的饱和蒸汽压。xA为溶液中A的摩尔分数。
.拉乌尔定律只适用于理想液态混合物或理想稀溶液中的溶剂。
2.亨利定律
pB=kx,BxB
式中kx,B为亨利系数,其值与溶质、溶剂的性质及温度有关。也可用其他浓度,如cB、bB来表示亨利定律,但这时亨利系数的大小及单位皆应相应改变。
此式只适用于理想稀溶液中的溶质。当挥发性溶液的浓度较大时,应以活度代替浓度,并要求溶质在气相中的分子形态与液相相同。
五、理想液态混合物
定义:其任一组分在全部组成范围内都符合拉乌尔定律的液态混合物。
pB=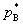 xB
xB
其中,0≤xB≤1,B为任一组分。
2.理想液态混合物中任一组分B的化学势
μB(l)=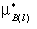 +RTln(xB)
+RTln(xB)
其中,为纯液体B在温度T、压力p的化学势。
若纯液体B在温度T、压力下的标准化学势为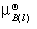 ,则有
,则有
=+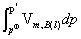 ≈
≈
其中,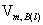 为纯液态B在温度T下的摩尔体积。
为纯液态B在温度T下的摩尔体积。
3. 理想液态混合物的混合性质
(1)△mix V=0
(2)△mix H=0
(3)△mix S=-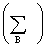 R
R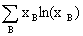
(4)△min G=—T△mix S
六、理想稀溶液
1.溶剂的化学势
μA(l)= +RTln(xA)
+RTln(xA)
 =
= + RTln(xA)+
+ RTln(xA)+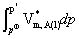
当P与相差不大时,积分项可忽略,则A的化学势为=+ RTln(xA)。稀溶液溶剂服从拉乌尔定律,溶质服从亨利定律,故稀溶液的溶剂化学势的表示与理想溶液中任一组分的化学势表达式一样。
2.溶质的化学势
溶质服从亨利定律,故
μB(溶质)=μB(g)=+ RTln=+ RTln(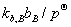 )
)
= + RTln(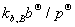 )+ RTln(
)+ RTln(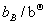 )
)
又因为+ RTln()=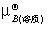 +
+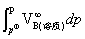
μB(溶质)=+ RTln( )+
)+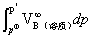
其中,在p与相差不大时,可忽略积分项。
3.分配定律
在一定温度、压力下,当溶质在共存的两不互溶溶液间平衡时,若形成理想稀溶液,则溶质在两液相中的质量摩尔浓度之比为一常数:
K=bB(α)/bB(β)
其中K为分配系数,bB(α)、bB(β)为溶质B在α、β两相中的质量摩尔浓度。
七、稀溶液的依数性
溶剂蒸气压下降:△pA=xB
凝固点降低(条件:溶质不与溶剂形成固态溶液,仅溶剂以纯固体析出):
△Tf=KfbB
Kf=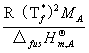
沸点升高(条件:溶质不挥发):
△Tb=KbbB
Kb=
渗透压:ⅡV=nBRT
八、逸度与逸度因子
1.逸度及逸度因子
气体B的逸度pB,是在温度T、总压力p总下,满足关系式
μB(g)=+ RTln
的物理量,它具有压力单位。其计算式为
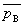 pB exp
pB exp
逸度因子(即逸度系数)为气体B的逸度与其分压之比
ΦB= /pB
理想气体逸度因子恒等于1。
2. 逸度因子的计算与普通化逸度因子图
lnΦB=
对纯真实气体,式中VB(g)即为该气体在T、p下的摩尔体积,用=ZRT/p代替VB(g)得lnΦ=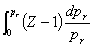
不同气体,在相同对比温度Tr、对比压力pr下,有大致相同的压缩因子Z,因而有大致相同的逸度因子Φ。
3.路易斯—兰德尔逸度规则
混合气体组分B的逸度因子等于该组分B在混合气体温度及总压下单独存在时的逸度因子。
=ΦB pB=ΦB p总y总=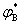 p总,y总=
p总,y总=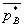 yB
yB
适用条件:由几种纯真实气体在恒温恒压下形成混合物时,系统总体积不变,即体积有加和性。
九、活度与活度因子
对真实液态混合物中的溶剂:
μB(l) +RTln(aB)= +RTlnxBfB
+RTln(aB)= +RTlnxBfB
其中aB为组分B的活度,fB为组分B的活度因子。
若B挥发,而在与溶液的气相中B的分压PB,则有
aB= pB/
且fB=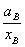 =
=
第五章 化学平衡
一、化学反应的等温方程
1.化学反应亲和势的定义
A=—△rGm
A代表在恒温、恒压和非体积功W=0的条件下反应的推动力,A>0反应能组分进行;A=0处于平衡态;A<0反应不能自发进行,反而是其逆反应会自发进行。
2.摩尔反应吉布斯函数与反应进度的关系
(аG/аξ)T, p= =△rGm
=△rGm
式中的(аG/аξ)T, p表示在T、p及组成一定条件下,反应系统的吉布斯函数随反应进度的变化率,称为摩尔反应吉布斯函数。
3.等温方程
△rGm=△r +RTlnJp
+RTlnJp
式中△r为标准摩尔反应吉布斯函数;Jp=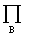 (pB/)vB。此式适用于理想气体或低压气体在恒温、恒压及恒组成的条件下,按化学反应计量式进行单位反应的吉布斯函数变的计算。
(pB/)vB。此式适用于理想气体或低压气体在恒温、恒压及恒组成的条件下,按化学反应计量式进行单位反应的吉布斯函数变的计算。
二、标准平衡常数
1.定义式
△r=— RTln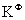
式中称为标准平衡常数;=Jp(平衡)。此式适用于理想气体或低压下气体的温度T下,按化学反应计量式进行 单位反应时,反应的△r与的相互换算。
二、理想气体反应系统的标准平衡常数
=(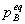 /)vB
/)vB
式中为化学反应中任一组分B的平衡分压。
3.有纯凝聚态物质参加的理想气体反应系统的标准平衡常数
= ((g)/)vB(g)
((g)/)vB(g)
三、温度对标准平衡常数的影响
化学反应的等压方程—范特霍夫方程
微分式 dln/dT=/RT2
积分式 ln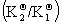 =(T2-T1)/( RT2T1)
=(T2-T1)/( RT2T1)
不定积分式ln=—/(RT)+C
对于理想气体反应,为定值,积分式或不定积分式只适用于为常数的理想气体恒压反应。若是T的函数,应将其函数关系式代入微分式后再积分,即可得到ln与T的函数关系式。
四、压力、惰性组分、反应物配比对理想气体化学平衡的影响
1.压力对平衡转化率的影响
= =
=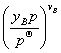 =
= ×
×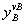
增高压力,反应向有利于体积减小的方向进行。
2.惰性组分对平衡转化率的影响
=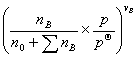 =
=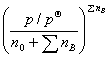 ×
×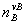
vB为参加化学反应各组分的化学计量数,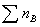 和
和 分别为对反应组分(不包括惰性组分)的物质的量、化学计量数求和。当 =0时,恒压下加入惰性组分对转化率无影响;当>0,恒压下加入惰性组分,平衡向生成产物的方向移动;若<0,则正好相反。
分别为对反应组分(不包括惰性组分)的物质的量、化学计量数求和。当 =0时,恒压下加入惰性组分对转化率无影响;当>0,恒压下加入惰性组分,平衡向生成产物的方向移动;若<0,则正好相反。
3.反应物的摩尔配比对平衡转化率的影响
对于气相化学反应aA+bB=yY+zZ
当摩尔配比r=nB/nA=b/a时,产物在混合气体中的含量为最大。
五、真实气体反应的化学平衡
=
 (/)vB =(
(/)vB =(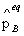 /)vB
/)vB
上式中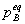 、
、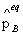 、
、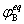 分别为气体在B化学反应达平衡时的分压力、逸度和逸度系数。则为用逸度表示的标准常数,有些书上用
分别为气体在B化学反应达平衡时的分压力、逸度和逸度系数。则为用逸度表示的标准常数,有些书上用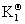 表示。
表示。
上式中=。
六、混合物和溶液中的化学平衡
1.常压下液态混合物中
=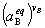 =
=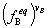 ×
×
2.常压下液态溶液中的化学平衡
=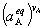 =
=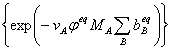 ×
×
第六章 相平衡
一、相率
F=C-P+2
其中,C=S-R- R`
式中F为系统的独立变量数,即自由度:C为组分数;R为独立的平衡反应数;R`为独立的限制条件数,即除了任一相中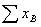 =1,同一物质在各平衡相中的浓度受化学势相等的限制以及R个独立化学反应的平衡常数对浓度的限制之外其它的浓度(或分压)限制条件皆包含于R`之中。S为系统的化学物质,P为相数。公式中的“2”代表T和p两个条件。此式只适用于只受温度、压力影响的平恒系统。
=1,同一物质在各平衡相中的浓度受化学势相等的限制以及R个独立化学反应的平衡常数对浓度的限制之外其它的浓度(或分压)限制条件皆包含于R`之中。S为系统的化学物质,P为相数。公式中的“2”代表T和p两个条件。此式只适用于只受温度、压力影响的平恒系统。
二、杠杆规则
杠杆规则在相平衡中是用来计算系统分成平衡两相(或两部分)时,两相(或两部分)的相对量。如图6-1所示,设在温度T下,系统中共存在的两相分别为α相与β相。图中M、α、β分别表示系统点与两相的相点;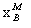 、
、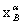 、
、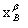 分别代表整个系统、α相和β相的组成(以B的摩尔分数表示);n、
分别代表整个系统、α相和β相的组成(以B的摩尔分数表示);n、 和
和 则分别为系统点、α相与β相的物质的量。由质量 得
则分别为系统点、α相与β相的物质的量。由质量 得
nα(-)=nα(-)
或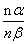 =
=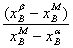

上式称为杠杆规则,它表示为α、β两相的物质的量的相对大小。如式中的组成由摩尔分数、、换成质量分数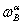 、
、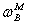 、
、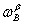 时,两相的量相应由物质的量nα与nα换成两相的质量mα与mα。
时,两相的量相应由物质的量nα与nα换成两相的质量mα与mα。
三、单组分系统相图
参考图6-2,单组分体系C=1,单相时,P=1则F=2,温度和压力均可变,称为双变量体系;汽化、凝固和升华时两相平衡(三条线上),P=2,则F=1,温度和压力只有一个可变,称为单变量体系;而其三相点(O点)外,P=3,则F=0,状态确定,温度、压力和组成都是定值,没有变量。
四、二组分图
1.气-液平衡相图
二组分体系C=2,若在恒温条件下,可作压力-组分图;若在恒压条件下,可作温度-组分图。此时自由度F=2-P+1=3-P。定压下,单项区F=2;二相区F=1;最高、最低恒沸点的混合物F=0。
参看图6-3,图(a)、(b)完全共溶系统气-液平衡的压力-组分相同。图(a)是A、B两种物质形成理想液态混合物的相图。图(b)是A、B两种物质形成真实液态混合物的相图,两图的差别很明显,图(a)的液相线为直线。图(c)是二组分理想液态混合物的气-液平衡温度-组成相图。图(d)是部分互溶系统的温度-组成相图,具有最低恒沸点的完全互溶体系与部分互溶体系的组合。图(e)是完全不互溶系统的温度-组成相图。由相图可以得出,共沸点低于每一种纯液体的沸点。
2.固-液相图
固相完全不互溶的固液平衡温度-组成图:图(a)是简单低共熔混合物;图(b)是形成稳定化合物;图(c)是形成不稳定化合物。
固相部分互溶的固液平衡温度-组成图:图(d)是体系具有低共熔点;图(e)是体系有转熔温度。
图6-4所示的二组分固-液相图具有以下共同特征:1)图中的水平线均是三相线;2)图中垂线都表示化合物。
五、三组分系统
利用相率研究体系中相和自由度的变化C=3,F=C-P+2=5-P,自由度最小为0,则相数最多是5;相数最少为1,自由度最多是4。
采用固定温度和压力,利用正三角形的三边表现三组分的摩尔分量,得到体系中各相得平衡曲线。此时条件自由度F*=3-P。
1.相图类型
相图类型(如图6-5所示)分为两大类:
(1)部分互溶的三液体系
有一对部分互溶的三液体体系(a)
有二对部分互溶的三液体体系(b)
有三对部分互溶的三液体体系(c)
(2)二固-液的水盐体系
无水合盐和复盐形成的体系;
有水合盐形成的体系(d)
有复盐形成的体系(e)
利用二固-液的水盐体系采用逐步循环法进行盐类的提纯和分离。
2.三组分体系相图的共同特征
(1)扇形区为固液平衡二相区。
(2)三角形区为三相区,各相成分和状态由三角形的顶端描述。
(3)杠杆规则适用于两相区,三相区接连适用两次杠杆规则同样可以确定各相的量,三组分相图用于材料特性(例如反磁性、超导性等)的研究。
第七章 电化学
一、法拉第定律
Q=Zfξ
通过电极的电量正比于电极反应的反应进度与电极反应电荷数的乘积。其中F=Le,为法拉第常数,一般取F=96485C·mol 近似数为965000C·mol。
二、离子迁移数及电迁移率
电解质溶液导电是依靠电解质溶液中正、负离子的定向运动而导电,即正、负离子分别承担导电的任务。但是,溶液中正、负离子导电的能力是不同的。为此,采用正(负)离子所迁移的电量占通过电解质溶液总电量的分数来表示正(负)离子导电能力,并称之为迁移数,用t+ ( t-) 表示,即
正离子迁移数
t+=Q+/(Q++Q-)=v+/(v++v-)=u+/(u++u-)
负离子迁移数
t_=Q-/(Q++Q-)=v-/(v++v-)=u-/(u++u-)
上述两式适用于温度及外电场一定而且只含有一种正离子和一种负离子的电解质溶液。式子表明,正(负)离子迁移电量与在同一电场下正、负离子运动速率v+ 与 v-有关。式中的u+ 与u- 称为电迁移率,它表示在一定溶液中,当电势梯度为1V·m-1 时正、负离子的运动速率。
其电解质溶液中含有两种以上正(负)离子时,则其中某一种离子B的迁移数计算式为
tBz+=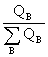
三、电导、电导率、摩尔电导率
1.电导
电阻的倒数称为电导,单位为S(西门子)。
G=1/R
2.电导率
电极面积为1 ,电极间距为1 时溶液的电导,称为电导率,单位为
G=1/R=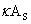 /l
/l
3.摩尔电导率
在相距为单位长度的两平行电极之间,放置有1 电解质溶液时的电导,称为摩尔电导率,单位是S·m2·mol-1。
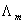 =
=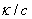
4摩尔电导率与电解质溶液浓度的关系式
(1)柯尔劳施(Kohlrausch)公式
= —A
—A
式中 是在无限稀释条件下溶质的摩尔电导率;c是电解质的体积摩尔浓度。在一定温度下对于指定的溶液,式中A和皆为常数。此式中适用与强电解质的稀溶液。
(2)柯尔劳施离子独立运动定律
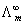 =v+
=v+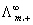 +v-
+v-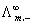
式v+ 及v- 分别为正、负离子的计量系数;及分别为在无限稀释条件下正、负离子的摩尔电导率。此式适用与一定温度下的指定溶剂中,强电解质或弱电解质在无限稀释时摩尔电导率的计算。
四、电解质的平均离子活度、平均离子活度因子及德拜—休克尔极限公式
1.平均离子活度
 ±
± (
(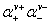 )
)
2.平均离子活度因子
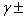 (
(
3.平均离子质量摩尔浓度
b± (b
(b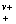 b
b )1/v
)1/v
4.离子活度
a=a =aa=
=aa= (b±/b
(b±/b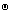 )
)
5.离子强度与德拜—休克尔极限公式
离子强度的定义式为
I=1/2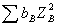
式中 bB 与 zB 分别代表溶液中某离子B的质量摩尔浓度和该离子的电荷数。I的单位为 mol·kg-1 。I值的大小反应了电解质溶液中离子的电荷所形成静电场强度的强弱。I的定义式用于强电解质溶液。若溶液中有强、弱电解质时,则计算I值时,需要将弱电解质解离部分离子计算在内。
德拜—休克尔极限公式为
lg =—Az+|z-|
=—Az+|z-|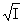
上式是德拜-休克尔从理论上导出的计算的式子,它只适用于强电解质极稀浓度的溶液。A为常数,在25℃的水溶液中A=0.509(mol-1·kg-1)-1/2。
五、可逆电池及其电动势
1.可逆电池热力学
(1) △rGm=Wr,m=-zFE
式中z是电池反应中电子转移数;F为法拉第常数;E是电动势。当△rGm<0 时,E>0 ,说明自发的化学反应恒温压下在原电池中可逆进行。
(2)△rSm=-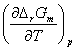 =zF
=zF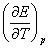
式中 称为原电池电动势的温度系数,表示恒温下电动势随温度的变化率,单位为
(3)△r H m=-z F E+zFT
(4)Qr,m= zFT
2.电动势的计算
(1)能斯特方程
化学反应为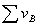 =0
=0
E=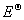 -
- ln
ln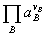
或E=- ln
ln
当电池反应达平衡时,△rGm=0,E=0,则
=ln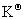
(2)电极的能斯特公式
E(电极)= 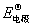 —ln
—ln
=+ln n
n
(3)任何可逆电池的电动势
E=E(右)-E(左)=E(阴)-E(阳)
= -
-
(4)液体接界电势
E(液界)=(t+-t-) ln
ln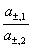
六、电极的种类
1.第一类电极
这类电极一般是将某金属或吸附了某种气体的惰性金属置于含有该元素离子的溶液中构成的,包括金属电极、氢电极、氧电极和卤素电极等。
2.第二类电极
第二类电极包括金属—难溶盐电极和金属—难溶氧化物电极。
3.氧化还原电极
任何电极均可发生氧化还原反应。这里所说的氧化还原电极专指如下一类电极:电极极板 只起输送电子的任务,参加电极反应的物质都在溶液中。如电极Fe3+, Fe2+ ;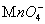 ,Mn2+,H+,H2O|Pt。
,Mn2+,H+,H2O|Pt。
七、极化电极电势
阳极:E(阳)=E(阳,平)+η(阳)η(阴)
阴极:E(阴)=E(阴,平)+η(阴)
式中 E(阳,平) 及 E(阴,平) 分别为阳极及阴极的平衡电板电势;η(阴)及η(阴)分别为阴、阳极的超电势。上述二式既适用与原电池,也适用于电解池个别电极的极化电极电势的计算。
第八章 量子力学基础
一、量子力学的基本假设
量子力学的4个基本假设是对3个问题的回答:一是运动状态如何描述;二是可观测的力学量如何表达;三是状态变化的规律。
1.波函数
由N个粒子组成的微观系统,其状态可由N个粒子的坐标(或动量)的函数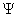 (t,q1,q2,…… ) 来表示, 被称为波函数。波函数是单值、连续的。
(t,q1,q2,…… ) 来表示, 被称为波函数。波函数是单值、连续的。
2.薛定谔方程
系统状态 (t,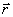 ) (代表所有坐标) 随时间的变化遵循薛定谔方程
) (代表所有坐标) 随时间的变化遵循薛定谔方程
-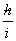
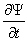 =
=
其中 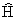 为哈密顿算符,=
为哈密顿算符,=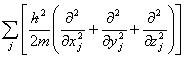 +V(t,)
+V(t,)
当势能与时间无关时,系统的波函数
(t,)=e-iEt/h()
3.系统所有可观测物理量的算符表示
量子力学中与力量学O对应的算符的构造方法:
(1)写出以时间、坐标和动量为坐标的力学量O的经典表达式
O(t;q1,q2,···;p1,p2,···)
式中 q1,q2,···表示动量 ; p1,p2,···表示坐标
(2)将时间t与坐标q1,q2,···看做数乘算符,而将动量pj 用算符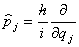 代替,则与力学量O对应的算符
代替,则与力学量O对应的算符 为
为
(t,q1,q2···;,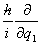 ,,
,,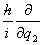 ,···)
,···)
4.测量原理
在一个系统中对力学量 进行测量的本征值 λn :
n=λnn
其有两层含义:
(1)如果系统所处的状态为 的本征态n ,则对 的测量结果一定为λn 。
(2)如果系统所处的状态 不是 的本征态,则对 的测量将使系统跃迁到 的某一本征态k ,其测量结果为该本征态对应的本征值λk 。
可将用 的本征态展开,即
=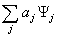
则测量结果为λk 的概率为 |ak|2。 一般来说,对处于状态 的系统进行测量,力学量 的平均值为
<> =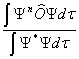
二、一维势箱中离子的薛定谔方程
-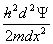 =E
=E
波函数(x)=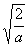 sin
sin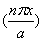 (n=1,2,3···)
(n=1,2,3···)
能及公式E=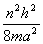 (n=1,2,3···)
(n=1,2,3···)
三、一维谐振子
哈密顿算符
=- +1/2kx2
+1/2kx2
能级Ev=(1/2+v)hv0
其中 v=0,1,2,3,··· 为振动量子数,v0=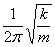 为谐振子经典基数。
为谐振子经典基数。
波函数
v=NvHv(ξ)exp(-ξ/2)
其中
ξ=x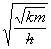 =x
=x
Nv=
Hv(ξ) 为 阶厄米多项式
Hv(ξ)=(-1)v exp(ξ2)( )
)
四、二体刚性转子
1.拉普拉斯算符在球级坐标中的表示
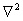 =
=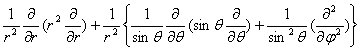
2.球谐函数
YJ,m( )=
)=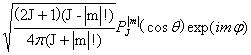
如设ξ=cosθ,则其中
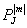 (ξ)=
(ξ)=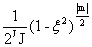
 (J
(J 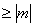 )
)
3二体刚性转子
若r 及V(r)均为常数,二体问题即成为二体刚性转子问题。若μ= m1m2/(m1+m2) ,则
EJ=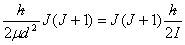 (J=0,1,2,···)
(J=0,1,2,···)
其中I=μd2 为转动惯量,波函数即为球谐函数YJ, m(θ, )
)
五、类氢离子及多电子原子的结构
1.类氢离子
V(r)=-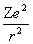
En=-
a0=0.5292×10-10m (n=1,2,3,······)
=Rn,J(r)·YJ, m(θ,)
其中:Rn,J(r)=—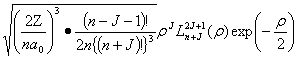
式中: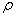 =
= 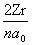 ,而
,而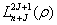 =
=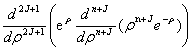

2.多电子原子
(1)多电子原子的哈密顿算符
=—
其中 =
= 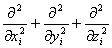 为第i个电子的拉普拉斯算符,ri 为它与核的距离,rij为电子i与电子j的距离,m为电子质量。
为第i个电子的拉普拉斯算符,ri 为它与核的距离,rij为电子i与电子j的距离,m为电子质量。
(2)多电子原子电子波函数
①中心立场近似法
将除电子i以外的其余Z-1个电子看做是球形对称分布的,电子i的势能为Vi=—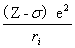 =—
=— 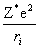 ,对不同电子σi 值不同。
,对不同电子σi 值不同。
=
n,J,m=R`(r)YJ,m(θ,)
En=—13.6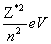
②自洽场方法
多原子的电子波函数为各个电子的波函数乘积:
(1,2···,Z)=
电子i与所有其他电子j的相互作用即为
Vij=e2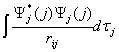
单电子哈密顿算符
 =—
=—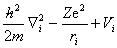
通过求解单电子薛定谔方程
(i)=
即可得到多电子薛定谔方程的解。可通过迭带过程求解。先假设一组单电子波函数。
3.斯莱特行列式
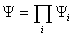 ,不满足费米子对波函数的反对称性的要求,斯莱特提出构造反对称波函数的一般方法。对N个电子的系统,若归一化的空间-自旋轨道组为{j ,j=1,2,3···} ,则反对称波函数表示为
,不满足费米子对波函数的反对称性的要求,斯莱特提出构造反对称波函数的一般方法。对N个电子的系统,若归一化的空间-自旋轨道组为{j ,j=1,2,3···} ,则反对称波函数表示为
(1,2···,N)=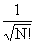
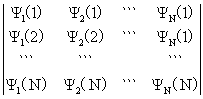

六、分子轨道理论简介
1.玻恩-奥本海默近似
分子系统中核的运动与电子的运动可以分离。
2.类氢分子离子的Schrodinger 方程的解
哈密顿算符
 =—
=—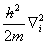 —
—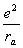 —
—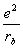
定义椭球坐标为
ξ= ,
,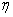 =
=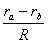
(1)Schrodinger 方程的解
el(ξ, ,)=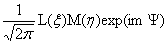 (m=0,±1,±2,···)
(m=0,±1,±2,···)
(2) 表8-1

对于坐标反演 (ξ, ,) (ξ, ,—+π) 波函数不变的用g表述,改变符号的用u表示。
(ξ, ,—+π) 波函数不变的用g表述,改变符号的用u表示。
(3)电子能级Eel(R)为核间距的函数,当核间距R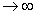 时趋于氢原子能级,核间距R 0时趋于氦正离子He+能级。
时趋于氢原子能级,核间距R 0时趋于氦正离子He+能级。
(4)U(R)= Eel(R)+e3/R 为势能曲线,对基态,在 R=Re=1.06×10-10m时有极小值—16.40eV。所以,该轨道为成键轨道。
第九章 统计热力学初步
一、离子各运动形式的能级及能级的简并度
1.三维平动子
εt=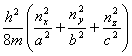 (nx,ny,nz=0,1,2···)
(nx,ny,nz=0,1,2···)
当a=b=c时有简并, (nx+ny+nz)相等的能级为简并的。
2.刚性转子(双原子分子)
εr =J(J+1)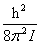 (J=0,1,2,···)
(J=0,1,2,···)
式中,I=μ ,μ=
,μ=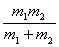 简并度为gr,J=2J+1。
简并度为gr,J=2J+1。
3.一维谐振子
εv=(v=1/2)hv (v=0,1,2···)
式中,v=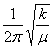 , k 为常数,μ为折合质量。能级为非简并的,即gv,v=1。
, k 为常数,μ为折合质量。能级为非简并的,即gv,v=1。
4.电子及原子核
系统中全部离子的电子运动及核运动均处于基态。电子运动及核运动基态的简并度为常数。分子能级是各种独立运动能级之和,为
ε=εt+εr+εv+εe+εn
二、能级分布微态数及系统总微态数
1.定域子系统
WD=N!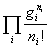
2.离域子系统
温度不太低时(即g i >>n i)WD=
一般情况下WD=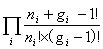
3.系统总微态数 =
=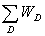
三、最概然分布与平衡分布
1.等概率定律
在N、U、V确定的情况下,假设系统各微态出现的概率相等。这个假设称为等概率定律。
P=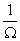
分布D出现的概率是
PD=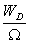
2.最概然分布和平衡分布
在N、U、V确定的条件下,微态数量大的分布称为最概然分布。而当N很大时,出现的分布方式几乎可以用最概然方式来代表。
N、U、V确定的系统平衡时,粒子的分布方式几乎不随时间而变化的分布,称为平衡分布。
四、玻耳兹曼分布
ni=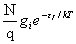
符合上式的分布称为玻耳兹曼分布。其中q为离子的配分函数。
q 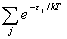 =
=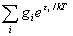
任两个能级上分布n i , n k之比为
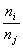 =
=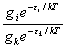
任一能级i上分布的粒子数 ni 与系统的总粒子数N之比为
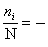
 =
=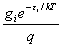
五、粒子配分函数的计算
1.配分函数的析因子性质
q = qt q r q v q e q n
2.能量零点的选择对配分函数的影响
若基态能级能量值为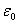 ,以基态为能量零点时,能量值
,以基态为能量零点时,能量值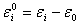
q = 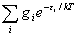 =
=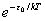
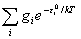 =
= q0
q0
即q0 = q
q
常温下,平动及转动配分函数与能量零点选择几乎无关,但振动配分函数与能量零点选择有关,即 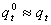
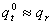
因为 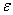 v,0 =hv/2
v,0 =hv/2
所以 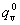 =qvexp(
=qvexp(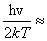 e5qv
e5qv
电子运动与核运动的配分函数与能量零点选择也无关。
3.配分函数的计算
(1)平动
 =
= 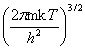 V
V
(2)转动(对线性刚性转子)
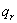 =
= 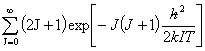
其中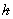 =
= 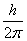
若设 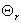 =
=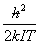 则当 T >> 时
则当 T >> 时

其中 为绕通过质心,垂直于分子的旋转轴一周出现的不可分辨的几何位置的次数,即分子对称数。对线性刚性转子转动自由度为2。
为绕通过质心,垂直于分子的旋转轴一周出现的不可分辨的几何位置的次数,即分子对称数。对线性刚性转子转动自由度为2。
(3)振动
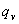 =
=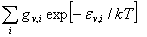 =e-hv/2kT
=e-hv/2kT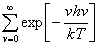
若设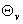 =
=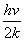 ,x=
,x=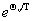 ,当 T<<
,当 T<< 时(常温),振动运动量子化效应突出,不能用积分代替加和;
时(常温),振动运动量子化效应突出,不能用积分代替加和;
=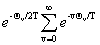 =
=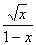
=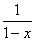
(4)电子运动
因为电子运动全部处于基态,电子运动能级完全没有开放,求和项中自第二项起均可忽略。所以

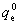 =
=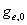 =Const
=Const
(5)核运动
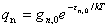
 =
=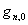 =Const
=Const
六、系统的热力学能与配分函数的关系
U i=NkT2
此处U i 可代表:
总热力学能。
零点为时的热力学能U0 = U — N
平动能,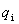 表示相应的配分函数。
表示相应的配分函数。
当U i 代表转动能、振动能、电子能、核能时,与V无关,偏微商可以写作全微商。
U i 与 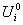 的关系:只有
的关系:只有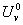 =
= ,其余:
,其余:




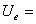
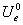
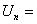

七、系统的摩尔定容热容与配分函数的关系
 =
=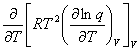 =
=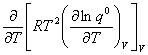
 ,与T无关,
,与T无关, 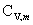 与零点能选择无关。
与零点能选择无关。
=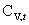 +
+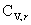 +
+
八、系统熵与配分函数的关系
1.玻耳兹曼熵定定理
S=kln
k为玻耳兹曼常数。
当N无限大时,最概然分布微态微 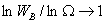 时,用
时,用 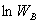 代替
代替 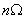 ,则 S=
,则 S=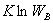 ,这种近似方法称为摘取量大项原理。
,这种近似方法称为摘取量大项原理。
2.熵与配分函数的关系
离域子系统:
S=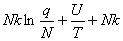 =
= 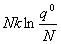 +
+
定域子系统:
S=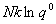 +
+ =
=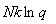 +
+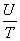
3.统计熵
通常把由统计热力学方法计算出系统的St 、Sr及Sv之和称为统计熵
S=St +Sr+Sv
Sm,t=R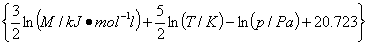 (理想气体)
(理想气体)
Sm,r=Rln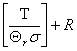
Sm,r=Rln(1- 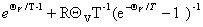 )
)
九、其他热力学函数与配分函数的关系
1.A、G、H与配分函数的关系
(1)A=-kTlnQ
Q=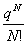 , A=—kT
, A=—kT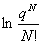 (离子域子系统)
(离子域子系统)
Q= 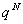 , A=—kT
, A=—kT (定域子系统)
(定域子系统)
(2)G=—kT+ NkTV (离域子系统)
(离域子系统)
G=—kT+ NkTV (定域子系统)
(3)H= NkT2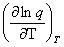 + NkTV
+ NkTV
2.理想气体的标准摩尔吉布斯函数
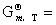 —RT
—RT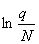 =—RT
=—RT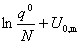
3. 理想气体的标准摩尔吉布斯自由能函数
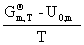 =—R
=—R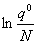
4.理想气体的标准摩尔焓函数
 =—RT
=—RT +R
+R
十、理想气体反应的标准平衡常数
— =
=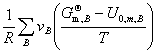 +
+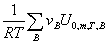
= —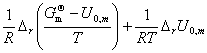
以平衡系统中各组分的粒子数 表示的平衡常数为
KN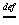
 =
=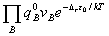
其中,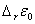 =
=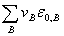
以平衡系统中各组分单位体积中的粒子数 表示的平衡常数为
KC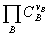 =
=
其中,分子浓度 CB NB/V
第十章 界面现象
一、表面功、表面吉布斯函数和表面张力
在温度、压力和组成不变的条件下,可逆地使表面积增加dAS 时,环境对体系所做的非体积功δ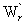 称为表面功,表示为
称为表面功,表示为
δ=γdAS
恒温、恒压下,可逆非体积功等于系统的吉布斯函数的增加,即
dGT,p=δ=γdAS
该式表明,若 dAS<0 ,则 dGT,p<0 ,即表面积减小的变化时自发的。
上式又可写作
γ=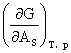
γ表示在单位面积上,表面层的分子比相同数量的内部分子多余的吉布斯函数,称表面吉布斯函数,其单位为J·m-2 。
γ又表示沿着液(或固)体表面并垂直作用在单位长度上的表面收缩力,称为表面张力,其单位为N·m-1 。
一、 弯曲液面的附加压力和蒸气压
1. 弯曲液面的附加压力
弯曲液面下的液体或气体均受到一个附加压力 的作用,该 的大小可由拉普拉斯方程计算,该方程为
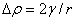
式中: 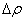 为弯曲液面内外的压力差;γ 为表面张力;r为弯曲液面的曲率半径。
为弯曲液面内外的压力差;γ 为表面张力;r为弯曲液面的曲率半径。
注意:(1)计算 时,无论凸液面还是凹液面,曲率半径r一律取正数,并规定弯曲液面的凹面一侧压力为p内,凸面一侧为p内 ,一定是p内 减p外 , 即
=p内 —p外
(2)附加压力的方向总指向曲率半径中心。
(3)对于在气相中悬浮的气泡,因液膜两侧有两个气液表面,所以气泡内气体所承受的附加压力为
=4γ/r
2. 弯曲液面附加压力引起的毛细现象
当液体润湿毛细管管壁时,则液体沿内管上升,其上升高度可按下式计算
h=2γcosθ/rg
式中:r 为液体表面张力; 为液体密度;g为重力加速度; 为接触角;r为毛细管内径。
注意:当液体不湿润毛细管时,则液体沿内管降低。
3. 微小液滴的饱和蒸气压——开尔文公式
RTln(pr/p)= 2γM/r
式中: pr 为液滴的曲率半径为r时的饱和蒸气压;p为平液面的饱和蒸气压; 、M 和γ分别为液体的密度、摩尔质量和表面张力。上式只用于计算在一定温度下,凸液面(如微小液滴)的饱和蒸气压随球形半径的变化。当计算毛细管凹液面(如过热液体中亚稳蒸气泡)的饱和蒸气压随曲率半径变化时,则上式的等式左边项要改写为RTln(p/ pr)。无论凸液面还是凹液面,计算时曲率半径均取正数。
一、 固体吸附
固体表面的分子由于受力不均而具有剩余力场,对气体分子产生吸引力,使气体在固体表面聚焦,从而降低固体的表面自由能,这种现象称为吸附。按吸附剂与吸附质作用本质的不同,吸附可分为物理吸附和化学吸附。
用单位质量吸附剂所吸附气体的物质的量n或其在标准状况下所占有的体积V来表示吸附量
na=n/m或Va=V/m
单位分别为mol·kg-1 或m3·kg-1。
1.朗缪尔单分子层吸附等温式
朗缪尔从吸附动态平衡的基本观点出发,提出了在均匀固体表面、吸附分子间无相互作用,只发生单分子层吸附情况下的吸附理论,推导出朗缪尔吸附等温式
θ=
式中θ为覆盖率,θ=Va/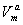 ,表示固体表面被覆盖的分数;b为吸附平衡常数,又称吸附系数,b值越大,则表示吸附能力越强;p为平衡时的气相压力。朗缪尔吸附等温式也可以表示为
,表示固体表面被覆盖的分数;b为吸附平衡常数,又称吸附系数,b值越大,则表示吸附能力越强;p为平衡时的气相压力。朗缪尔吸附等温式也可以表示为
Va=
式中 表示吸附搭饱和时的吸附量;Va则表示覆盖为θ时的平衡吸附量。当压力很低或吸附较弱时,bp《1 ,则上式可简化为 Va=bp;当压力足够高时或吸附较强时,bp》1
则上式可简化为 Va=。
2.吸附热力学
吸附式一个自发过程,是吉布斯函数下降的过程,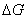 =
= 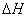 —T
—T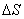 <0 。因吸附过程中,气体分子由三维空间被吸附到二维表面,自由度减小,<0 ,则 <0 。吸附通常为放热过程。
<0 。因吸附过程中,气体分子由三维空间被吸附到二维表面,自由度减小,<0 ,则 <0 。吸附通常为放热过程。
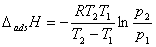
p1和 p2 分别是在 T1 和 T2 下达到某一个相同吸附量时的平衡压力。温度升高时,要想维持同样的吸附量,必然要增大气体的压力,即若T2>T1 ,必然 p2>p1。
四、液—固体面
1.接触角与杨氏方程
当一液滴在固体表面上不完全展开时,在气、液、固三相汇合点,液—固界面的水平线与气—液界面切线之间通过液体内部的夹角 ,称为接触角。其角度大小取决于同时作用于O处的液体分子之上的固体表面张力γs 、液体界面张力γls以及液体表面张力γl 。当平衡时,存在以下关系:
γs=γls+γlcosθ
以上公式只使用于光滑的表面。
2.湿润与铺展
湿润是固体表面上的气体被液体取代的过程。按湿润程度的不同,分为沾湿、浸湿和铺展3种。θ<90。的情形称为湿润;θ>90。的称为不湿润;θ=0。或不存在时称为完全湿润;θ=180。 时称为完全不湿润。
铺展是少量液体在固体表面上自动展开,形成一种薄膜的过程。用铺展系数S作为衡量液体在固体表面能否铺展的判据。
S=—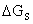 =γs-γls-γl
=γs-γls-γl
S≧0时,可发生铺展,S越大,铺展性能越好;S﹤0,则不能铺展。
五、溶液表面的吸附
1. 溶液表面的吸附现象
溶液的表面张力随溶质的性质及浓度而变化,所以溶液会自动调节表面层的浓度而尽量降低表面自由能,导致溶液表面层的组成与本体溶液的组成不同,称这种现象为溶液表面的吸附作用。若溶质在表面层的浓度大于它在本体溶液中的浓度,则为正吸附,反之为负吸附。
2.吉布斯吸附等温式
吉布斯吸附公式描述了描述了溶质的表面吸附量 与溶质的活度和表面张力随溶质活度变化率之间的关系:
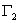 =—
=—
对于稀溶液,可用溶质的浓度代替活度,并可略去下角标,表示为
 =—
=—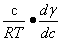
若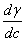 <0得>0,表明凡增加浓度使表面张力降低的溶质在表面层发生正吸附;
<0得>0,表明凡增加浓度使表面张力降低的溶质在表面层发生正吸附;
若>0得<0,表明凡增加浓度使表面张力增大的溶质在表面层发生负吸附。
3.表面活性剂
凡融入某液体后能使某液体的表面张力显著降低,在液体表面产生正吸附的物质称为表面
活性剂。
按化学结构来分类,大体上可分为离子型和非离子型两大类。
表面活性剂物质的基本性质包括外表面定向排列和内部形成胶束,它们都能降低表面张力。
表面活性剂物质在溶液中开始形成胶束的最低浓度为临界胶束浓度。
第十一章 化学动力学
一、化学反应速率及速率方程
1.反应速率
单位时间单位体积内化学反应的反应进度为反应速率。
v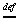 (1/vBV)(dnB/dt)
(1/vBV)(dnB/dt)
反应速率的单位为mol·m-3·s-1。其与用来表示速率的物质B的选择无关,与化学计量式的写法有关。
对于恒容反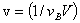
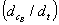
对于化学计量反应


 —···
—···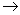 ···+
···+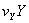 +
+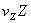
经常指定反应物A的消耗速率 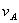 = —
= —
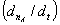 或某指定产物Z的生成速率
或某指定产物Z的生成速率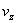 =
= 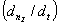 来表示反应进行的速率,则
来表示反应进行的速率,则
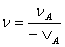 =
=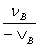 =···
=··· =
=
各不同物质的消耗速率或生成速率与各自的化学计量数的绝对值成正比。
对于恒温恒容气相反应,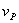 =
= 
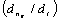 (恒容)
(恒容)
=vRT
2. 基元反应的质量作用定律
对基元反应 aA + bBP
其质量作用定律表示为 r=k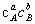
该式表示基元反应的速率与所用反应物浓度(带相应指数)的乘积成正比,其中浓度指数恰是反应式中各相应物质计量系数的绝对值。其中,比例系数k为基元反应的速率常数。
3. 反应速率方程
表示化学体系中反应速率与反应物的浓度间函数关系的方程式称为反应速率方程。由实验数据得出的经验速率方程一般可表示为
=— =k
=k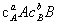 ···
···
式中nA 、nB 等分别称为反应组分的反应分级数;n= nA +nB +···为反应的总级数。反应级数的大小表示浓度对反应速率影响的程度,级数越大,则反应速率受浓度的影响越大。
二、具有简单级数反应的速率方程及特点

三、速率方程的确定
确定反应级数的三种常用方法:微分法、尝试法和半衰期法,后两种属于积分法。
1. 微分法
—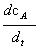 =k
=k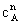 是速率方程的微分式,应用此式求反应级数的方法即为微分法。
是速率方程的微分式,应用此式求反应级数的方法即为微分法。
2. 尝试法
试差法又称为尝试法。就是看某一化学反应的cA与t间的关系适合于哪一级数的动力学积分式,从而确定该反应的反应级数。
3. 半衰期法
半衰期法确定反应级数的依据是化学反应的半衰期和反应物初始浓度之间的关系与反应级数有关。
四、反应速率与温度的关系
阿伦尼乌斯公式
指数式 k=A·e 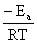
式中 为活化能;A为指前因子(GB3102—93称“指前参量”)。
为活化能;A为指前因子(GB3102—93称“指前参量”)。
对数式 lnk=+lnA
微分式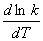 =
=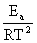
定积分式 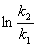 =
=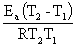
五、典型复合反应
1. 对行反应
如以正、逆反应均为一级反应。
A B
B
t=0  0
0
t=t  —
—
t=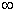
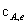 —
—
— =
=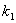 -
- 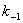 (-)
(-)
—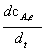 =-(-)=0
=-(-)=0
Kc=/=(-)/
—d(-)/dt =(+)(-)
当 Kc 很大,即 》 , 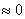 时
时
—=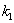
2. 平行反应


当两个反应都是一级反应,则
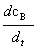 =k1
=k1
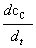 =k2
=k2
若反应开始时,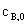 =
=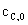 =0,则 +
=0,则 +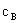 +
+ = ,所以
= ,所以
—=(+ 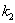 )
)
积分得ln(/)=(+ ) t
平行反应的特点:当组成平行反应每一个反应级数均相同时,则各个反应物的浓度比等于各反应的速率常数之比,而与反应物的起始浓度及时间无关。
3. 连串反应
假设由两个一级反应组成的连串反应。
A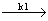 B
B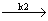 C
C
t=0 0 0
t=t
— =
=
—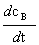 =-
=-
因为++=,则cC=
六、复合反应速率的近似处理法
1. 选取控制步骤法
连串反应的总速率等于最慢一步的速率。最慢的一步称为反应速率的控制步骤。控制步骤的反应速率常数越小,其他各串联步骤的速率常数越大,则此规律就越准确。这时,要想使反应加速进行,关键就在于提高控制步骤的速率。
2. 平衡态近似法
对于反应处理
A + B C (快速平衡)
C k2D (慢)
k2D (慢)
若最后一步为慢步骤,因而前面的对行反应能随时近似维持平衡。从化学动力学角度考虑,上面的快速平衡时正向、逆向反应速率近似视为相等。
3. 稳态近似法
在连串反应中
ABC
若中间物B很活泼,极易继续反应,则必k2》k1 。就是说第二步反应比第一步反应快的多,B一旦生成,就立即经第二步反应掉,所以反应系统中B基本上没什么积累,cB 很小。这时B的浓度使处于稳态或定态。所以稳态或定态就是指某中间物的生成速率与消耗速率相等以致其浓度不随时间变化的状态。
七、基元反应速率理论
1.简单碰撞理论
(1)双分子气体反应的碰撞数 (碰撞频率)
同种分子
异种分子
(2)双分子气体反应速率
同种分子
异种分子
其中 为 能。
2.过渡状态理论(活化络合物理论)
(1)浓度 为标准时的速率常数
其中 为玻耳兹曼常量,h为普朗克常量,n为所用反应物系数之和。
(2)对气相反应,以 为标准态时的速率常数
(3)溶液中离子反应速率的原盐效应
其中zA 、zB 为离子A、B的电荷;I为离子强度;k0为各种离子和活化络合物的活度系数均为1时的速率常数;25℃ 的水溶液A=0.509。
八、量子效率与量子产率
量子效率
量子产率
第十二章 胶体化学
一、胶体系统特点
分散相粒子在某方向上在1-1000mm范围的高分散系统称为胶体。可分溶胶、高分子溶液、缔合胶体3类。具有可透明或不透明性,但均可发生光散射,胶体粒扩散速率慢,不能透过半透膜,具有较高的渗透压的特点,其主要特征是高度分散的多相性和热力学不稳定性。
二、光学性质
当将点光源发出的一束可见光照射到胶体系统时,在垂直于入射光的方向上可观察到一个发亮的光锥,此现象称为丁铎尔现象。丁铎尔现象产生的原因是胶体粒子大小小于可见光的波长,而发生光的散射的结果。散射光的强度I可由瑞利公式计算:
I=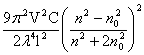 (1+cos2α)I0
(1+cos2α)I0
式中:I0及λ表示入射光的强度与波长;n及n0 分别为分散相及分散介质的折射率;α为散射角,即观测方向与入射光之间的夹角;V为单个分散相粒子的体积;C为分散相的数密度;l 为观测者与散射中心的距离。此式适用于粒子尺寸小于入射光波长,粒子看成点光源,而且不导电,还有不考虑粒子的散射光相互发生干涉。
三、胶体系统的动力性质
1.布朗运动
胶体粒子由于受到分散介质分子的不平衡撞击而不断地做不规则的运动,称此运动为布朗运动。其平衡位移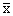 可按下列爱因斯坦-布朗位移公式计算
可按下列爱因斯坦-布朗位移公式计算
=(RTt/3Lπrη)
式中:t为时间,r为粒子半径, η为介质的粘度。
2.扩散、沉降及沉降平衡
扩散:指当有浓度梯度存在时,特质粒子(包括胶体粒子)因热运动而发生宏观上的定向迁移现象。
沉降:指胶体粒子因受重力作用而发生下沉的现象。
沉降现象:当胶体粒子的沉降速率与其扩散速率相等时,胶体粒子在介质的浓度随高度形成一定分布并且不随时间而变,这一状态称为胶体粒子处在沉降平衡。其数密度C与高度h的关系为
Ln(C2/C1) =—(Mg/RT)[{1- }(h2-h1)]
}(h2-h1)]
式中:  及
及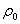 分别为粒子及介质的密度,M为粒子的摩尔质量,g为重力加速度。此式适用于单级分散粒子在重力场中的沉降平衡。
分别为粒子及介质的密度,M为粒子的摩尔质量,g为重力加速度。此式适用于单级分散粒子在重力场中的沉降平衡。
四、胶体的电学性质
胶粒表面电荷来源于电离作用、吸附作用和摩擦带电荷等。
施特恩(Stern)双层模型表示为图12-1所示。若固体表面带正电荷,则双电层的溶液一侧由两层组成,第一层是吸附在固体表面的反离子(与固体表面所带电荷相反),称为紧密层,第二层为扩散层。固体表面与溶液本体之间的电势差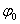 称为热力学电势;紧密层与扩散层的分界处同溶液本体之间的电势差
称为热力学电势;紧密层与扩散层的分界处同溶液本体之间的电势差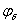 称为施特恩电势;滑动面与溶液本体之间的电势差称为
称为施特恩电势;滑动面与溶液本体之间的电势差称为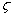 电势(亦称电动电势)。
电势(亦称电动电势)。
1.电动现象
在外电场的作用下,胶体粒子在分散介质中定向移动的现象,称为电泳。
若在多孔膜(或毛细管)的两端施加一定电压,分散介质将通过多孔膜而定向流动,称为电渗。
在外力的作用下,迫使分散介质通过多孔隔膜(或毛细管)定向流动,多孔隔膜两端所产生的电势差(它是电渗的逆现象)称为流动电势。
分散相粒子在重力场或离心力场的作用下迅速移动时,在移动方向的两端所产生的电势差(它是电泳的逆现象)称为沉降电势。
2.胶团的结构
胶团的结构(以KI过量的AgI溶胶为例)可表示为:
胶核一般选择吸附与胶核同组分的离子或能与胶核表面反应生成难溶物的物质。
五、溶胶的稳定与聚沉
溶胶稳定的原因有3个:胶体粒子带电、溶剂化作用和布朗运动。
聚沉:是指溶胶中胶粒互相聚集结成大颗粒,直到最后发生沉淀的现象。导致溶胶聚沉的因素很多,但电解质加入进溶胶发生沉淀的作用是显著的,为比较不同电解质对溶胶聚沉作用的大小而引进聚沉值,聚沉值是指令溶胶发生明显的聚沉所需的电解质最小浓度。聚沉值的倒数值称为聚沉力。
应指出:起聚沉作用的主要是与胶粒带相反电荷的离子(即反离子),反离子价数越高则聚沉值越小。离子价数及个数均相同的不同反离子,其聚沉能力亦不相同,如
H2>Cs+>Rb+>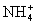 >K+>Na+>Li+
>K+>Na+>Li+
F—>Cl—>Br—> NO3—> I—> SCN—> OH—
-
大学物理化学知识点归纳
第一章气体的pvT关系一理想气体状态方程15MmixmnmBnBBBpVmMRTnRT1611式中MB为混合物中某一种组分B或pV…
-
大学有机化学知识点总结与归纳
1.酸性比较α-H存在于羰基的α-C上(这是句废话)它的酸性由旁边的吸电子基团的强弱决定一般的,相邻基团的吸电子效果越强其酸性越强…
-
天津大学物理化学知识点归纳
第一章气体的pvT关系一理想气体状态方程pVmMRTnRT11或pVmpVnRT12式中pVT及n的单位分别为Pam3K及molV…
-
大学物理化学上册知识点归纳
第一章气体的pvT关系一理想气体状态方程15MmixmnmBnBBBpVmMRTnRT1611式中MB为混合物中某一种组分B或pV…
-
大学有机化学复习总结
有机化学复习总结一有机化合物的命名命名是学习有机化学的语言因此要求学习者必须掌握有机合物的命名包括俗名习惯命名系统命名等方法要求能…
-
大学有机化学复习总结
有机化学复习总结一、有机化合物的命名命名是学习有机化学的“语言”,因此,要求学习者必须掌握。有机合物的命名包括俗名、习惯命名、系统…
-
大学有机化学知识点总结与归纳
1.酸性比较α-H存在于羰基的α-C上(这是句废话)它的酸性由旁边的吸电子基团的强弱决定一般的,相邻基团的吸电子效果越强其酸性越强…
-
大学有机化学期末复习总结
本课程的学习即将结束,现将全书的重点内容按命名、结构理论、基本反应、化合物转化及合成方法、鉴别等几个专题进行总结归纳,供同学们复习…
-
大学有机化学期末复习总结
有机化学复习资料公共基础学院一楼文印室提供价格实惠欢迎您光临!一、有机化合物的命名命名是学习有机化学的“语言”,因此,要求学习者必…
-
大学有机化学期末复习总结
有机化学复习资料公共基础学院一楼文印室提供价格实惠欢迎您光临!有机化学复习总结一、有机化合物的命名命名是学习有机化学的“语言”,因…
-
初三下学期物理化学知识点大总结
初三下学期物理化学知识点大总结.txt22真诚是美酒,年份越久越醇香浓型;真诚是焰火,在高处绽放才愈是美丽;真诚是鲜花,送之于人手…