无机化学 知识点总结
第一章物质存在的状态………………………………………………………………2
一、气体... 2
二、液体... 3
①溶液与蒸汽压... 3
②溶液的沸点升高和凝固点的下降... 3
③渗透压... 4
④非电解质稀溶液的依数性... 4
三、胶体... 4
第二章 化学动力学初步……………………………………………………………5
一、化学反应速率... 5
二、化学反应速率理论... 6
三、影响化学反应速率的因素... 6
2、温度... 7
第三章 化学热力学初步……………………………………………………………8
一、热力学定律及基本定律... 8
二、化学热力学四个重要的状态函数... 9
4、自由能... 10
①吉布斯自由能... 10
②自由能G——反应自发性的判据... 11
③标准摩尔生成自由能 ............ 11
............ 11
④有关 的计算... 11
的计算... 11
三、化学热力学的应用... 11
一、化学平衡... 13
二、化学平衡常数... 13
无机化学(上) 知识点总结
第一章 物质存在的状态
一、气体
1、气体分子运动论的基本理论
①气体由分子组成,分子之间的距离>>分子直径;
②气体分子处于永恒无规则运动状态;
③气体分子之间相互作用可忽略,除相互碰撞时;
④气体分子相互碰撞或对器壁的碰撞都是弹性碰撞。碰撞时总动能保持不变,没有能量损失。
⑤分子的平均动能与热力学温度成正比。
2、理想气体状态方程
①假定前提:a、分子不占体积;b、分子间作用力忽略
②表达式:pV=nRT;R≈8.314kPa·L·mol ·K
·K
③适用条件:温度较高、压力较低使得稀薄气体
④具体应用:a、已知三个量,可求第四个;
b、测量气体的分子量:pV= RT(n=)
RT(n=)
c、已知气体的状态求其密度ρ:pV=RT→p= →ρ
→ρ =p
=p
3、混合气体的分压定律
①混合气体的四个概念
a、分压:相同温度下,某组分气体与混合气体具有相同体积时的压力;
b、分体积:相同温度下,某组分气体与混合气体具有相同压力时的体积
c、体积分数:φ=
d、摩尔分数:xi=
②混合气体的分压定律
a、定律:混合气体总压力等于组分气体压力之和;
某组分气体压力的大小和它在混合气体中体积分数或摩尔数成正比
b、适用范围:理想气体及可以看作理想气体的实际气体
c、应用:已知分压求总压或由总压和体积分数或摩尔分数求分压、
4、气体扩散定律
①定律:T、p相同时,各种不同气体的扩散速率与气体密度的平方根成反比:
 =
= =
= (p表示密度)
(p表示密度)
②用途:a、测定气体的相对分子质量;b、同位素分离
二、液体
1、液体
①蒸发气体与蒸发气压
A、饱和蒸汽压:与液相处于动态平衡的气体叫饱和气,其气压叫做饱和蒸汽压
简称饱和气;
B、特点:a、温度恒定时为定值;
b、气液共存时不受量的变化而变化;
c、物质不同,数值不同
②沸腾与沸点
A、沸腾:当温度升高到蒸汽压与外界压力相等时,液体就沸腾,液体沸腾时的温度叫做沸点;
B、特点:a、沸点的大小与外界压力有关;外界压力等于101kPa时的沸点 为正常沸点;b、沸腾是液体表面和内部同时气化的现象
2、溶液
①溶液与蒸汽压
a、任何物质都存在饱和蒸汽压;
b、纯物质的饱和蒸汽压只与物质本身的性质和温度有关;
c、一定温度下饱和蒸汽压为常数;
d、溶液蒸汽压的下降:△p=p -p
-p =K·m
=K·m
②溶液的沸点升高和凝固点的下降
a、定量描述:沸点升高 △T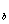 =K·m
=K·m
凝固点下降 △T =K·m
=K·m
仅适用于非电解质溶液
b、注 意:①T、T的下降只与溶剂的性质有关
②K、K的物理意义:1kg溶剂中加入1mol难挥发的非电解质溶质时,沸点的升高或凝固点下降的度数
c、应用计算:i、已知稀溶液的浓度,求△T、△T
ii、已知溶液的△T、△T求溶液的浓度、溶质的分子量
d、实际应用:i、制冷剂:电解质如NaCl、CaCl
ii、实验室常用冰盐浴:NaCl+HO→22°C
CaCl+HO→-55°C
iii、防冻剂:非电解质溶液如乙二醇、甘油等
③渗透压
a、渗透现象及解释:
渗透现象的原因:半透膜两侧溶液浓度不同;
渗透压:为了阻止渗透作用所需给溶液的额外压力
b、定量描述:Vant'Hoff公式:
∏V=nRT ∏= 即∏=cRT
即∏=cRT
∏为溶液的渗透压,c为溶液的浓度,R为气体常量,T为温度。当浓度c较小时,可近似为c≈m
④非电解质稀溶液的依数性
a、难挥发非电解质稀溶液的蒸汽压下降、凝固点下降、沸点上升和渗透压变化都与溶液中所含的种类和性质无关,只与溶液的浓度有关,总称溶液的依数性,也叫非电解质稀溶液的通性。
b、注意:上述非电解质稀溶液的有关计算公式用于电介质稀溶液时要乘以相应电解质中溶液中的质点数;但浓溶液不能用上述公式计算。
三、胶体
1、胶体的组成:分散相+分散介质+稳定剂
2、胶体的性质:
①光学性质:丁达尔效应————胶团对光的散射现象;
②动力性质:布朗运动—————胶团粒子的不规则运动;
③电学性质:电泳现象—————胶粒在电场下的不规则运动
3、溶胶的稳定性
①动力学稳定性:胶团运动
②聚集稳定性:胶粒的带电性使同种电荷有排斥作用;
③热力学稳定性:胶体粒子因很大的比表面积而能聚集成大颗粒
4、胶体的聚沉———关键:稳定性的去除
①加电解质,如明矾使水净化(吸附电荷);
②与相反电性的溶胶混合;
③加热
第二章 化学动力学初步
一、化学反应速率
①表达:化学反应速率可用反应物或生成物的浓度随时间的变化率来表示。
②数学表达式:对于反应A→B:
 =
= 或
或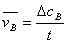
注:以反应物浓度减少和生成物浓度增大和生成物浓度增大表示是符号不同;用不同物质浓度来表示反应速率不同。
2、反应进度
①定义:对于化学计量方程式,若定义d ,称
,称 为反应进度。表示物质变化量除以相应的计量系数。
为反应进度。表示物质变化量除以相应的计量系数。
②表达式: ,
, 表示化学计量系数。
表示化学计量系数。
③表式意义:表示一个反应进行的程度;其纲量为摩尔;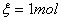 指按化学计量方程式进行一个单位的反应
指按化学计量方程式进行一个单位的反应
④注 意:反应进度的表示与计量方程式的写法有关。
3、速率方程和速率常数
①速率方程:把反应物浓度和反应速率联系起来的数学表达式。
对于反应:aA+Bb→gG+hH
反应速率v=k·c (A)·c
(A)·c (B),即为速率方程式,式中的常数k即为反应速率常数。
(B),即为速率方程式,式中的常数k即为反应速率常数。
②反应速率常数:
a、物理意义:k只取决于反应的本性(E ,活化能)和温度;
,活化能)和温度;
b、注意事项:k是温度的函数,与浓度的大小无关;
k的单位即量纲,随速率方程变化而变化;
k一般由实验测得,只有基元反应可以直接写出。
③速率方程的实验测定
作图法:由浓度—时间动力学曲线可得到斜率k及速率常数;
初速法:可得到个反应的反应级数
4、基元反应和非基元反应
①基元反应:反应物分子在有效碰撞过程中经过一次化学变化就能转化为产物的反应;
注 意:由一个基元反应构成的化学反应又称简单反应;
只有基元反应才能根据质量作用定律直接写出速率方程
②非基元反应:反应分子需经过几步反应才能转化为反应产物的反应。
注 意:非基元反应的速率方程不能根据反应式写出速率方程,必须根据实验测定的结果有反应历程推出,并验证;
复杂的非基元反应→分成若干个基元反应→最慢一步发宁作为苏空反应步骤
5、反应级数
①定义:速率方程中各反应物浓度的指数;
②说明:如v=k·c 则反应物A的反应级数为m,反应物B的反应级数为n;总反应级数为m+n
则反应物A的反应级数为m,反应物B的反应级数为n;总反应级数为m+n
③注意:a、反应级数表示了反应物浓度对反应速率影响的大小关系;反应级数只能由实验测定;
b、反应级数可以是整数、分数、零或负数;
c、零级反应的反应速率与反应物浓度无关
④反应级数的确定
基本方法
a、测定反应物浓度c随时间t的变化;
b、作c-t图像,求个时刻的速度v;
c、分析v与浓度c的变化关系,确定m、n
二、化学反应速率理论
1、碰撞理论
①主要内容:反应物分子间的相互碰撞是反应进行的必要条件,反应物分子碰撞频率越高,反应速率越快,但并非每次碰撞都能引起反应发生,能发生化学反应的碰撞为有效碰撞
②有效碰撞发生的条件:
a、相互碰撞的分子应有适合的碰撞取向;
b、相互碰撞的分子必须具有足够的能量。把能够发生有效碰撞的分子称为活化分子
③根据碰撞理论,增大化学反应速率的方法:
a、增大单位时间内分子碰撞的总数————增大浓度;
b、增大碰撞总数中有效碰撞的百分数———升高温度
④活化能:碰撞理论认为,活化能是活化分子的平均能量与反应物分子的平均能量之差
2、过渡态理论
①主要内容:化学反应并不是通过简单碰撞就能完成的,而是在反应物到生成物的过程中 经过一个高能的过渡态,处于过渡态的分子叫做活化络合物。活化络合物是一种是一种高能量的不稳定的反应物原子组合体,它能较快的分解为新的能量较低的生成物。
②活化能E:过渡态理论认为,活化能是反应物分子能量与处于过渡态的活化络合物分子的平均能量之差
3、活化能:决定反应速率的内在因素
①活化能在一定温度范围内可认为是常数;
②活化能对反应速率的影响很大;E越小,反应速率越大;
③催化剂可以改变反应的活化能,故可以降低化学反应速率
三、影响化学反应速率的因素
1、浓度:由速率方程v=k·c 知,浓度对化学反应速率有一定的影响
知,浓度对化学反应速率有一定的影响
压强对化学反应速率与的影响是通过浓度来实现的。
2、温度
①范特霍夫规则:对于一般的化学反应,温度每升高10K,反应速率增加2-4倍
②阿伦尼乌斯公式:
a、表达式: ,其中,A为特征常数,既指前因子;E为经验常数即活化能,k为反应速率常数,R为摩尔气体常数8.314
,其中,A为特征常数,既指前因子;E为经验常数即活化能,k为反应速率常数,R为摩尔气体常数8.314 ,e为自然对数底,该公式的对数形式为
,e为自然对数底,该公式的对数形式为
b、应用:(1)求某一温度下某反应的k:
作图法:lgk对 作图可得一直线关系;斜率:
作图可得一直线关系;斜率: ;截距lg斜率大的活化能E大,反应速率随温度的升高增加较快
;截距lg斜率大的活化能E大,反应速率随温度的升高增加较快
二点法:不同温度下反应速率常数k的计算
(2)由lgk-的图像得出的结论:
i、同一反应,低温低和高温时变化同样的温度,低温时反应速率变化大;即一个反映在低温时速率随温度变化比高温区更显著
ii、不同反应,变化相同的温度时,E大的反应k变化大。升高温度有利于 大的反应
大的反应
3、催化剂及基本特征
a、催化剂和催化作用:正催化剂;负催化剂(阻化剂)
b、催化剂的特征:i、催化剂只改变反应速率,不改变反应方向;
ii、催化剂同等程度地改变正逆反应的活化能,同时提高正逆化学反应速率;
iii、催化剂具有一定的选择性;
iv、催化剂在反应前后不发生变化,但在反应过程中会变化
第三章 化学热力学初步
一、热力学定律及基本定律
1、基本概念
①环境与体系
a、体系
i、定义:人为划出的作为研究对象的一部分空间。
ii、分类:敞开体系(与外界可进行物质能量交换)
封闭体系(只有能量交换)
孤立体系(物质、能量均不与外界交换)
b、环境:出体系以外的其他部分,与体系存在能量交换
②功和热:
a、热(Q):i、系统与环境由于温差而传递的能量
Q﹥0,体系从环境中吸热;Q﹤0,体系从环境中放热;
物体之间可通过功、热、辐射三种形式交换能量
ii、热容、比热容、摩尔热容
b、功:i、热力学中除热外,其它各种被传递的能量统称为功
气体膨胀做功:W=-pΔV
环境对体系做功,W﹥0;体系对环境做功,W﹤0
③状态及状态函数
a、状态:体系的某种存在状况。它由一系列的物理量决定,如气体的p、V、T 等,一旦体系处于一定的状态,体系的所有其它性质都有确定值
b、 状态函数:在特定状态下,某一性质具有唯一值,则称该状态为状态函数。
c、结论:状态一定值一定,殊途同归变化同,周而复始变化零。
④过程与途径
a、过程:体系变化状态变化的经过
b、途径:变成一个过程所经历的具体步骤
c、注意:体系状态函数的变化只取决于体系始终变化的过程,而与变化的路径无关
⑤广度性质与强度性质
a、广度性质:及容量性质,与体系中物质的量成正比的量,具有加和性
b、强度性质:数值上不随体系中物质总量的变化而变化的物理量,不具加和性
⑥热力学标准态:
当系统中各种气态物质的气压均为标准压力 ,固态和液态物质表面承受的压力等于压力,溶液中各物质的浓度均为1
,固态和液态物质表面承受的压力等于压力,溶液中各物质的浓度均为1 时,我们就说物质处于热力学标准态。
时,我们就说物质处于热力学标准态。
注:热力学标准态并未对温度有限制,热和温度都有热力学标准态。
2、热力学定律:
①热力学第一定律
a、实质:能量守恒与转化定律
b、数学表达式:
c、注意:i、Q与W的符号;ii、功和热不是状态函数,但两者之和是状态函数
②热力学第二定律:揭示了宏观过程的方向与限度
熵增加原理:孤立体系有自发向混乱度增加的方向变化的趋势
③热力学第三定律:任何纯物质的完整晶体在T=0K时的熵值为零
二、化学热力学四个重要的状态函数
1、热力学能(内能)
①符号U,是系统内各种形式能量的总和
②内能的变化△U
a、对于孤立体系,环境改变,内能不变;对于非体系,
b、标准摩尔反应热力学能变化(反应内能变化):符号△ ,表示反应是在热力学标准态下进行的,其值的大小与化学反应方程式的书写一一对应。
,表示反应是在热力学标准态下进行的,其值的大小与化学反应方程式的书写一一对应。
2、焓
①焓
a、定义:H≡U=pV
b、焓变:
c、符号规定:放热反应,△H﹤0;吸热反应,△H﹤0
d、单位:kJ·mol
e、表式意义:化学反应在等温等压下发生,不做其它功时,化学反应的热效应等于系统状态函数的变化量
f、注意:△H表示每摩尔反应即时,而不是每摩尔反应物
②热化学方程式
a、表式意义:表示化学反应与其热效应关系的化学方程式
b、标准写法(以下列为例):
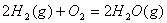


c、书写化学方程式的注意事项
i、需注明反应条件;
ii、需注明各物质的存在状态;
iii、正确表达反应的热效应
d、标准反应焓
定义:当反应物和生成物都处于标准态时的反应焓变,用来表示
非标准态(压力不为)时,则表示为
单位: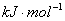 ,标准状态下,反应进度为时的焓变
,标准状态下,反应进度为时的焓变
3、熵
①混乱度和熵
混乱度:即无序度,其大小与体系中存在的微观粒子数目有关
熵:体系混乱度的热力学度量
表示符号:S;单位: ;熵是状态函数,具有广度性质
;熵是状态函数,具有广度性质
②绝对熵:任何温度下的熵值,及温度为T时,
注:绝对熵为相对值
③反应熵:发生1mol时熵变化的简称,
化学反应的上增大还是减小,有时很容易判断:
凡气体分子增多的反应,一定是熵增反应;反之是熵减小反应;反应前后气体分子数目不变的反应难以熵增还是熵减
④标准熵
a、定义:在标准状态下,1mol纯净物质的绝对熵叫标准熵
b、符号: ;单位:
;单位: ;注:应指定温度T
;注:应指定温度T
c、熵的可比性
i、在相同条件下, ﹥
﹥ ﹥
﹥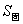 ;
;
ii、结构相似,相对分子量不同的物质, 随相对分子量的增加而增大
随相对分子量的增加而增大
iii、相对分子量接近,分子结构越复杂的,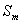 也越大
也越大
⑤标准摩尔反应熵变
a、热力学标准态下,反应终始态物质的熵值之差
b、标准摩尔反应熵变(标准反应熵):
c、意义:i、正值(熵增加)倾向自发过程;
ii、负值倾向非自发过程
4、自由能
①吉布斯自由能
a、定义:封闭系统在等温等压条件下向环境可能做的最大有用功对应的状态函数,符号为 ;
;
b、计算公式:
在等温条件下可以理解为,焓变(△H)=自由能变化(△G)+热力学温度(T)×熵变(△S)
c、意义:当△G﹤0时, ﹥0,表明自由能被用来做最大有用功,是自发过程;
﹥0,表明自由能被用来做最大有用功,是自发过程;
当△G﹥0时,﹤0,表明过程非自发进行,必需由环境对体系做功
②自由能G——反应自发性的判据
定温定压下,任何自发变化总是反应吉布斯自由能函数减少
 G﹤0,自发过程,正向反应自发;
G﹤0,自发过程,正向反应自发;
G﹥,非自发过程,其逆反应为自发过程
③标准摩尔生成自由能
定义:在热力学标准态下你,稳定单质的生成自由能为零。由稳定单质生成1mol纯物质时反应的自由能变化为该物质的标准摩尔生成自由能。
符号: 单位:
标准状态下,稳定单质的=0
④有关的计算
方法一:
已知标态下的, ,求
,求
当温度变化不大时,可近似认为是常数,为298K时的值
方法二:由求  (生成物)
(生成物)
方法三:根据盖斯定律求
方法四:对的修正公式
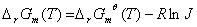

三、化学热力学的应用
1、盖斯定律
①内容:如果一个反应可以分几步进行,则总反应的焓变等于各分步反应焓变之和,前提是保持反应条件(温度、压力)不变。
②应用:用于进行太慢或反应速率不易控制而无法直接测得反应热的化学反应。
③应用条件:a、某化学反应是在等压(或等容)的条件下进行的,在分步完成时各分步反应也要在等压(等容)条件下进行
b、要消去某一物质时,不仅要求物质的种类相同,其物质的聚集状态也要相同。
第四章 化学反应的热平衡
一、化学平衡
1、化学平衡的建立:在一定的条件下,可逆化学反应的正负反应速率相等时的状态。
2、化学平衡的特点:①是一个动态的过程;②在平衡状态下, ;③化学平衡的组成与到达平衡的途径无关。
;③化学平衡的组成与到达平衡的途径无关。
3、注意:化学平衡是相对的、有条件的、暂时的动态平衡;
从热力学(宏观)上讲,在一定条件下达到平衡状态时,系统的吉布斯自由能达到最低时的状态,,在宏观上表现为静止;
从动力学(微观)上分析,处于平衡的反应不过是正逆反应速率相等时的状态,是整个反应保持平衡,实际上正逆反应仍在进行
二、化学平衡常数
1、标准平衡常数
①定义:在一定的温度下,可逆反应达到平衡时,以产物计量系数为幂的平衡浓度的乘积与以反映计量系数为幂的平衡浓度的乘积的比值为常数,即化学反应的标准平衡常数,用符号 表示。
表示。
②表达式:对于反应 ,
,  ,量纲为1
,量纲为1
③平衡常数的意义:化学平衡常数是反应进行程度的标志。值越大,反应进行程度越大,反应进行得越彻底。
④影响因素:的大小与反应物自身的性质和反应的温度有关,而与体系中各组分的浓度、分压无关
⑤书写标准平衡表达式的注意事项
a、式中各组分浓度或分压为反应平衡时的浓度或分压;
b、反应中有固体和纯液体物质时,其浓度视为常数,不写在表达式中;对于非水溶液,当水作为溶质时,其浓度应视为常数,写在表达式中;
c、的表达式与方程式的书写有关;
d、多重平衡原则:总反应的等于各相加分反应的之积;正逆反应的标准平衡常数互为倒数
e、平衡常数的大小与温度有关,除298K温度外,在其它状态下,书写时标明温度
⑥有关计算:已知反应的 求平衡转化率
求平衡转化率
2、实验平衡常数
①定义:实验得到的平衡常数叫做实验平衡常数或经验平衡常数。
②分类:浓度平衡常数;分压平衡常数
③表达式:浓度平衡常数表达式 
压强平衡常数表达式 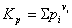
二者关系:
④与标准平衡常数的比较
a、标准煤平衡常数的量纲统一为1;
b、实验平衡常数有 之分,且量纲不统一,特殊情况下才为1;
之分,且量纲不统一,特殊情况下才为1;
3、反应熵J(分压熵 或浓度熵
或浓度熵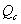 )
)
①表达式:对于反应
浓度商
压强商
②与平衡常数的比较
J﹥K时, ﹥0,反应逆向进行;
﹥0,反应逆向进行;
J=K时,=0,反应达到平衡;
J﹤K时,﹤0,反应正向进行
三、化学平衡的移动
1、平衡移动:从旧的平衡状态转移到新的平衡状态的过程
2、勒沙特列原理:一旦改变维持化学平衡的条件,平衡就会向着减弱这个改变的方向移动。
注意:①该原理不能实际判定某一系统是否达到平衡,只是预言了平衡打破后,体系的新的平衡移动的方向;
②平衡移动的结果是建立了新的平衡体系;
③利用勒沙特列原理不能进行定量计算
第五章 酸碱平衡
一、酸碱理论介绍
①酸碱的概念:凡是在水溶液中电离产生的阳离子全部是 的物质叫做酸;凡是电离产生的阴离子全部是
的物质叫做酸;凡是电离产生的阴离子全部是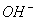 的物质叫做碱;在水溶液中电离产生的阳离子除外尚有其它离子或电离产生的阴离子除外尚有其它离子的物质的物质叫做盐。
的物质叫做碱;在水溶液中电离产生的阳离子除外尚有其它离子或电离产生的阴离子除外尚有其它离子的物质的物质叫做盐。
②中和反应的实质:
③局限性:a、该理论的立论基础是水溶液中电解质的电离,不能脱离水溶液的范畴,非水体系不适用;
b、只适用于含、的物质,无法解释 、
、 呈碱性的事实;
呈碱性的事实;
2、布伦斯惕——劳伦酸碱质子理论
①酸碱的概念:凡是能给出的物质都是酸;凡是能接受的物质都是碱
②共轭酸碱对:质子酸碱不是孤立的,它们通过质子相互联系,质子酸释放质子转化为它的共轭碱,质子碱得到质子转化为它的共轭酸。这种关系称为酸碱共轭关系,其酸碱称为共轭酸碱对。
③酸碱反应
实质:质子转移的过程;
中和反应的实质:质子的传递;
电离作用的实质:水与分子酸碱之间的质子传递;
阿伦尼乌斯酸碱理论中盐类水解的实质:水与离子酸碱之间的质子传递
④注意:a、在质子酸碱理论中,酸和碱可以是分子,可以是离子;
b、同一物质有时可以做酸,有时可以做碱;
c、质子理论中没有盐的概念,也无水解反应;
d、单独一对共轭酸碱对本身是不能发生酸碱反应的
3、路易斯酸碱理论
二、水的电离和pH标度
1、水的离子自递
①水即是质子酸又是质子碱,水分子之间可以进行质子传递:

简写为 ,即为水的电离
,即为水的电离
②水的离子积
纯水的电离平衡常数称为质子自递常数,简称为水的离子积。
298K时,纯水的 ,
,
水的质子自递是吸热反应,故 随温度的升高而增大
随温度的升高而增大
2、水溶液的pH值
①溶液的pH值 
方便起见,当的浓度小于1时,用pH表示溶液的酸度;当溶液的浓度大于1时,用的浓度,表示溶液的酸度。
 也可以表示溶液的酸碱度,
也可以表示溶液的酸碱度,
 298K时,
298K时,
另外,水溶液的酸碱性取决于与的浓度的相对大小
[]﹥[]时,溶液呈酸性;[]﹤[]时,溶液呈碱性;[]=[]时,溶液呈中性
②酸碱测定计
③生物学意义
④酸碱指示剂
假设In表示石蕊,HIn(红)

当c(HIn)﹥﹥c(In )时,溶液呈红色,是酸性;
)时,溶液呈红色,是酸性;
当c(HIn)﹤﹤c(In)时,溶液呈蓝色,是碱性
甲基橙变色范围:---红---3.1---橙---4.4---黄---
酚酞变色范围:---无色---8.0---粉红---9.8---红---
三、水溶液中的酸碱电离平衡
1、电离度与稀释定律:
①电离度(α):已电离的弱电解质分子数和溶液中弱电解质分子初始分子数的百分比:
②稀释定律:在一定温度下,( 为定值),某弱电解质的电离度随溶液浓度的减小而增加
为定值),某弱电解质的电离度随溶液浓度的减小而增加
2、酸碱的强弱
①电离平衡常数
对于任意酸,达到电离平衡时,

为弱酸电离平衡常数,用来衡量酸的强弱,值越大,酸性越强
﹥1时,为强酸;﹤1时为弱酸
多元弱酸对应多级电离平衡常数
同理,对于弱碱来讲, 越大,碱性越强
越大,碱性越强
注意:酸碱的电离平衡常数表征了酸碱给出质子或接受质子的能力
②共轭酸碱与的联系:=
③区分效应与拉平效应
a、区分效应:溶剂使酸的强度得以显出差别的效应称为区分效应,该溶剂被称为区分溶剂。
b、拉平效应:溶剂将酸的强度拉平的效应称为拉平效应,该溶剂称为拉平溶剂。
④、酸碱强度大小与:a、酸碱本身:b、溶剂有关
3、同离子效应和盐效应
①同离子效应:在弱电解质电离平衡体系中,加入和弱电解质含有相同离子的强电解质会使弱电解质的电离度减小的现象。
②盐效应:向弱电解质溶液中加入强电解质会使弱电解质电离度增大的现象。
③注意:同离子效应一定会产生盐效应,盐效应不一定会产生同离子效应
4、相关计算
①一元弱酸或弱碱的电离平衡;
②多元弱酸或弱碱的电离平衡
5、盐类水解
①盐类水解与水溶液的相互作用:
强酸强碱盐——无水解作用;
强酸弱碱盐、弱酸强碱盐、弱酸弱碱盐——有水解作用
②水解反应
a、定义:指盐的组分离子与水电离出来的或结合生成电解质的过程
b、实质:中和反应的逆反应
③类型
A、强酸弱碱盐电离的实质——弱酸的电离
以 为例:
为例:
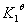


总: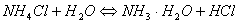


溶液水解后产生,因此溶液呈酸性,溶液pH可按一元弱酸处理
B、弱酸强碱盐电离实质——弱碱的电离
以 为例:
为例:

总:

溶液中水解后产生,因此溶液呈碱性,溶液pH可按一元弱碱处理
C、弱酸弱碱盐电离的实质——两性物质电离
以 为例
为例 


总:

水解后产生和,溶液的酸碱性由、的相对大小决定
④影响盐类水解的因素
a、盐的浓度:同一温度下,盐的浓度越小,水解程度越大;
b、温度:水解反应为吸热反应,温度升高,水解程度增大;
c、溶液的酸碱性及同离子效应。
⑤注意 :有些盐类,如 、
、 可以完全水解
可以完全水解
四、缓冲溶液
1、缓冲溶液的定义、组成、原理
①缓冲溶液的定义:缓冲溶液是外加酸碱pH变化很小的溶液
②缓冲溶液的组成:弱酸及其盐组成的溶液,弱碱及其共轭酸组成的溶液,多元弱酸弱碱的共轭酸碱对组成的溶液。
③缓冲溶液的原理:同离子效应,电离平衡的移动
2、缓冲容量和缓冲范围
①缓冲容量:缓冲能力大小的衡量,指单位体积缓冲溶液的pH值改变极小值所需的酸或碱的物质的量。
②有效缓冲范围:缓冲溶液具有稳定溶液pH值的能力,但这种能力是有限的,即有效缓冲范围。
如果外加酸碱过多,超过缓冲范围,缓冲溶液会失去缓冲能力。
3、影响缓冲溶液的因素
①缓冲溶液的pH主要由 或14-决定
或14-决定
②缓冲对的浓度:缓冲容量与体系共轭缓冲对的总浓度有关。总浓度越大,缓冲容量越大。
③缓冲对的浓度之比:当c(酸)/c(碱)=1时,缓冲容量最大。
④有效缓冲范围:缓冲对浓度之比在10-1/10范围内,有较好的缓冲能力。
即最有效的缓冲范围一般在 或
或
4、缓冲溶液的选择与配制
①选择:
a、所选择的溶液除了参与、的有关反应外,不能与反应系统中的其他物质发生副反应 ;
b、所选的弱酸的或弱碱的尽可能的接近缓冲溶液的pH;
c、一般要求缓冲溶液的组分浓度为0.05--0.5,缓冲溶液有足够的缓冲能力;
d、缓冲溶液廉价易得,避免污染。
②配制
a、根据要求选择缓冲对;
b、计算缓冲物质的浓度之比,使所配溶液的pH为所需值:

c、根据计算结果配制缓冲溶液,并使酸碱浓度均在0.1--1之间;
d、用酸度计或试纸测定所配溶液的pH
5、缓冲现象的应用
①自然界与生物界
②用标准缓冲溶液标定其他物质的酸度
③许多反应要求反应在一定范围内进行
第六章 沉淀溶解平衡
一、沉淀溶解平衡常数——溶度积
1、沉淀溶解平衡:一定温度下,难溶电解质变成水合离子的(即溶解)的速率与溶液中水合离子回到晶体表面(即沉淀)的速率相等时的状态叫做沉淀溶解平衡。
2、溶度积
①定义:一定温度下,难溶电解质的饱和溶液中各离子浓度的系数方次的乘积,记为 。
。
②表达式:对于反应 ,
,
③注意:a、溶度积是难溶电解质饱和溶液的特性常数;
b、属于化学平衡常数,其大小只与难溶电解质的自身性质和温度有关,与溶液中离子的浓度无关。
c、适用范围:难溶强电解质及难容弱电解质
3、溶度积与溶解度S
①相同点:都可以反应难溶电解质的溶解能力的大小;
②不同点:a、溶解度是平衡常数的一种,表示一定温度下难溶电解质的饱和溶液中离子浓度之间的关系;
b、溶解度S是浓度的一种表达形式,表示一定温度下1L难溶电解质中溶质的量;
③注意:比较不同电解质溶解度时,若电解质属于相同类型,则可直接比较,大的,S也大;比较不同类型的电解质溶液时应先利用求出溶解度S,然后比较大小
二、沉淀的生成与溶解
1、离子积:①定义:某难溶电解质溶液中,其离子浓度系数方次之积,用J( )表示。
)表示。
②注意:J与表达式相同,但概念不同,其关系如同反应商与标准平衡常数之间的关系。
2、溶度积规则
①依据:
推论:



②对沉淀溶解与生成的的判断
J﹤,﹤0,不饱和溶液,无沉淀生成,有沉淀溶解质饱和;
J﹥,﹥0,过饱和溶液,有沉淀生成,至达到饱和;
J=,=0,饱和溶液,处于沉淀溶解平衡状态。
3、同离子效应和盐效应
①同离子效应:向难溶电解质中加入含有相同离子的强电解质难溶电解质的沉淀---溶解平衡向生成沉淀的方向移动,使其溶解度减小。这种现象称为同离子效应。
②盐效应:在难溶电解质中加入易溶强电解质而使难溶电解质的溶解度增大的作用。
③注意事项:a、当加入可溶性的含相同离子的电解质时,会产生一定的盐效应。但所加的可溶性盐浓度较小时,同离子效应占主导因素;当浓度较大时,盐效应对沉淀溶解平衡的影响大于同离子效应;
b、有同离子效应必然会产生盐效应,而盐效应不一定会产生同离子效应
4、分步沉淀与沉淀的转化
①分步沉淀:溶液中同时含有几种离子,由于它们的溶解度不同,形成沉淀所需离子的浓度不同,加入同一种电解质,溶解度小的先从溶液中沉淀出来。
②离子沉淀的先后顺序取决于:
a、 沉淀物的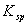 :同类型的难溶电解质,当被沉淀的离子的浓度相同时,越小的越先被沉淀出来;
:同类型的难溶电解质,当被沉淀的离子的浓度相同时,越小的越先被沉淀出来;
b、 被沉淀离子的浓度:不同类型难溶电解质或者被沉淀离子的浓度不同时,则不能比较,要具体计算,根据溶解度才能判断
③沉淀的转化:在含有沉淀的的溶液中加入适当的沉淀剂,与溶液中的某一离子结合成为更难溶的电解质,从而从一种沉淀转化成为了另一种沉淀。
同一类型难溶电解质在同一温度下值越大越容易转化
三、沉淀反应的应用
1、除杂
①通过控制pH值除去某些金属离子;
②利用金属硫化物沉淀反应除去 、
、 、
、 等;
等;
③工业上除食盐中可溶性杂质 、
、 、
、
2、离子鉴定
3、离子分析
4、分析化学
①重要分析法
②滴定分析法
第七章 电化学基础
一、离子-电子法配平氧化还原方程式
1、配平原则
①反应过程中氧化剂所获得的电子数必须等于还原及时取得电子数;
②反应前后各元素的原子总数相等
2、配平步骤
①将分子反应式改为离子反应式;
②将离子反应式分成两个为配平的半反应式,半反应式两边的原子数和电荷数相等。如果半反应两边的的氢、氧原子个数不相等,则按反应的酸碱条件,在酸性介质中添加 或
或 ,在碱性介质中添加
,在碱性介质中添加 或;
或;
③用左右两边添加电子使半反应两边的电荷数相等的办法配平板反应方程式;
④根据电子得失求出最小公倍数,将两个半反应分别乘以相应的系数,使反应中得失电子总数相等,然后将两个半反应相加,同时注意未变化的离子的配平,并恢复成分子方程式。
注意:离子-电子法配平氧化还原方程式只适用于水溶液的氧化还原反应
二、原电池
1、原电池(Primary cell)
①几个基本概念
电极: 电子产生与流动的源泉
导线: 电子流过的外电路
盐桥: 离子流过电解质溶液的通路
②电极
正极: 发生还原反应, 例铜正极半反应: Cu2+ + 2e = Cu
负极: 发生氧化反应, 例锌负极半反应: Zn = Zn2+ + 2e
电极反应--正、负电极的半反应、电池反应--半反应之和
③原电池符号及写法: (-) Zn|ZnSO4 ( c )‖CuSO4( c )|Cu (+)
负极 界面(s/l) 盐桥 界面 正极
2、电极电势(electrode potential)
①形成: 溶解(溶剂化)Û沉积
溶解(溶剂化)Û沉积
②概念:金属在其盐溶液中达到溶剂化离子平衡时, 金属表面内侧与外侧(溶液)之间产生的电势差。
③物理意义:电极电势是金属在溶液中失去电子能力大小的一种量度。
电极电势的表示: -
-
④影响因素
A、电极的本性:电极电势取决于金属的活泼性。金属越活泼,电极电势越低。反之,电极电势越高。
B、金属离子的浓度
C、溶液的温度
⑤电池电动势:
化学电池的电动势是电池反应的化学驱动力
⑥标准氢电极
A、标准氢电极的构造与原理
国际统一规定---
298K时标准氢电极的电极电势规定为 0 V,即
B、应用:由此可求得常见电对的标准电极电势
C、标准电极电势? ? (Standard electrode potential)
在热力学标准状态下,某电极的电极电势称为该电极的标准电极电势? ?
D、测量:在热力学标准状态下,给定电极与标准氢电极(指定为负极)组成原电池,测得原电池的电动势E
3、原电池的最大功和吉布斯自由能
根据吉布斯自由能的定义知,在恒压等温条件下,当体系发生变化时,体系吉布斯自由能等于对外所做的最大非体积功,用公式表示为 ;
;
如果非体积功只有电功一种,则上式又可写为
式中,n为电池输出电荷的物质的量,单位为mol;E为可逆电池的电动势,单位为V;F为法拉第电常数, 。
。
三、能斯特方程
1、电极电势的能斯特方程式
非标态下的电极电势可用能斯特方程式求出:
注意:①电极反应式中的反应系数为能斯特公式中各物质浓度的方次;
②电极反应式中的固体和纯液体不写入公式,其它物质均要表现在式中,溶液用浓度表示,气体则要用分压与标准压强的比值;
③公式中的氧化型和还原型物质不只包括氧化数有变化的物质
2. 电极电势的影响因素
①电对中氧化型或还原型物质的浓度的改变
氧化型浓度↑,电对的 ↓;还原型的浓度↑,电对↓ ?
↓;还原型的浓度↑,电对↓ ?
②溶液酸碱性对电极电势的影响
对于有或参加的电极反应,溶液的酸碱性对氧化还原产物也有影响
③生成沉淀或配合物对电极电势的影响
实质:氧化型或还原型的离子浓度降低,从而使电对的电极电势降低或者升高
四、电极电势的应用
1、计算原电池的电动势:
2、判断氧化剂或还原剂的相对强弱 :电极电势的值的高低可以判断氧化剂或还原剂的相对强弱;E的值越高,电对的氧化态是越强的氧化剂;E值越低,电对的还原剂是越强的还原剂。
3、判断氧化还原进行的方向
原电池电动势E与自由能变化ΔG的关系
对于可逆电池,等温等压下体系自由能的减少等于原电池所作的最大电功
数学表达式:
n: 反应中由还原剂转移到氧化剂的总电子数;
F: 法拉第常数( 或)
或)
标准状态下则:
化学反应方向的判断对于E >0和ΔG<0是一致的
由 可知
可知
已知组成氧化还原反应的两个电对的?,
? ﹥
﹥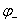 ,E>0,ΔrGm <0,反应正向;
,E>0,ΔrGm <0,反应正向;
?=,E=0,ΔrGm =0,反应达到平衡;
?<,E<0,ΔrGm >0,反应逆向;
在标准状态下,则用 或
或 来判断方向。
来判断方向。
当两个电对的相差不大时,浓度将对反应方向起决定作用;
当两个电对相差即电动势大于0.2V时,一般浓度变化不会影响反应的方向
4. 判断标准态下氧化还原反应的程度
电池电动势与标准平衡常数 的关系
的关系
由公式和 推出
推出
在298K时,
一般来讲≥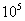 表示反应程度较大,可以认为反应相当完全。相当n=1时,=0.3V;
表示反应程度较大,可以认为反应相当完全。相当n=1时,=0.3V;
注意:应用
已知反应的电动势求反应的和
已知反应的电动势求各物质的平衡浓度
5. 测定溶度积常数和稳定常数
根据Nernst公式,通过测定原电池的电动势或直接根据电对的电极电势可求得难溶性强电解质的溶度积常数和配离子的稳定常数 ;
;
6. 判断氧化还原反应进行的次序
根据,与氧化剂电对差值越大的还原剂电对的还原剂越先被氧化
四、元素电势图及歧化反应
1、概念:以图解方式表示某一元素不同氧化态物种之间组成电对的电极电势关系的方法称为元素电势图。
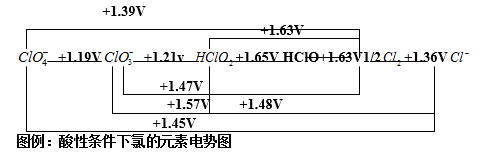
注意:物种氧化态由左到右降低,氧化态标在各物种下方;
横线上方数值为相邻两物种构成电对的电极电势值;
同一元素有酸、碱性元素电势图两种,一般要注明(A,B)
2、元素电势图的应用
由已知电对求未知电对的标准电极电势
由于电极电势是电极的强度性质不具有加合性,因此要通过该电极反应的标准自由能变化来计算。
计算公式: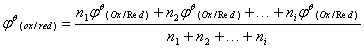
(Ox/Red):未知电对的标准电极电势;
1(Ox/Red)、2(Ox/Red)……i(Ox/Red):分别为相邻电对的标准电极电势;
n1、n2……ni:分别为相邻电对的电极反应中电子转移的个数。
判断岐化反应(disproportionation)和反岐化反应(comproportionation)发生的可能性
岐化反应的概念:中间氧化态的物种反应生成高、低氧化态的物种。例:
Br2 + 2OH- = Br- + BrO- + H2
(氧化数) 0 -1 +1
判断依据:在元素电势图中某一中间氧化态物种左边的?Θ小于右边的?Θ,则该物种在水溶液中可发生岐化反应;反之左右两侧物种则发生反岐化反应。
注意:元素电势图一定要表明酸碱条!
第八章 原子结构
一、原子结构
1、基本结构:由电子、中子、质子构成
关键问题:电子排布及化学性质之间的关系
2、原子
① 电子和原子核:带正电,原子核电子之间有静电吸引;形成化学键时电子运动发生改变,原子核不变
② 核的结构:带正电的质子和不带电的中子;质子与中子的强吸引作用与质子间静电作用的相互对抗。Z增加排斥作用增强,排斥作用占主导。稳定元素的数目有限。
③ 核素:具有一定质子数和一定中子数的的原子的总称。
④ 同位素:质子数相同而中子数不同的一类原子的总称。天然同位素组成元素,原子量由同位素的比例定。化学性质非常相似。
⑤ 同位素丰富度:某元素的各种天然同位素的分数(原子百分比)称为丰富度。
⑥ 放射性:不稳定的核因发射高能粒子而分解。Z﹥83(Bi)的元素都具有放射性。
⑦ 原子质量:以u为单位
⑧ 原子的相对原子质量(原子量): 核素原子量的1/12
核素原子量的1/12
二、波尔行星模型与氢原子结构的量子力学模型
1、氢原子光谱
①实验规律: ,n=1,2,3,…
,n=1,2,3,… 为Hydberg常数,数值为
为Hydberg常数,数值为
②原子光谱的特点:
A、不连续性;B、谱线波长之间具有一定简单的数学关系。
2、波尔理论
①基本假设
a、行星模型:波尔的氢原子模型可形象地称为行星模型
b、定态假设:核外电子具有一定的轨道;稳定态(定态、激发态)——与电磁学说相反
c、量子化条件:
 →氢原子的基态能量为-13.6eV(n=1)
→氢原子的基态能量为-13.6eV(n=1)
d、跃迁规则:电子可在不同轨道间跃迁,
结论:对于氢原子,计算值与实际值惊人的吻合,误差值小于0.1%,波尔成功地解释了氢原子光谱,上升为波尔理论。
不连续性-Rutherford原子模型与Maxwell电磁理论的矛盾;
②波尔理论的局限性
优点:
冲破了经典力学中能量连续的束缚,引入量子化概念、跃迁规则;给出了玻尔理论中的几个核心概念:定态、激发态、跃迁、能级;
缺点:
1). 未能完全冲破经典物理的束缚,仍使用行星模型的固定轨道,仍用离心力=向心力的传统牛顿力学!忽视了电子运动的一个重要特征-波粒二象性;
2). 不能解释多电子原子的光谱学规律。
解决之道:完全冲破经典的牛顿力学束缚,建立新体系(量子力学),引入新概念(波粒二象性)。
3、波粒二象性
德布罗意的波粒二象性假设:假设光具有二象性,那么微观粒子在某种条件下也应具有波动性。
能量 动量
动量
D、 P→粒子性; →波动性
→波动性
德布罗意关系式: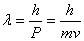
4. 测不准原理(Uncertainty principle)
海森堡认为: 不可能同时精确测得电子的位置和动量(速率) 。
测不准关系式: ,
, →位置不确定量;
→位置不确定量;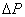 →动量不确定量
→动量不确定量
经典力学中,物体有精确的轨道(轨迹),并在某一瞬间有准确的速率。
新的量子力学中,微观粒子的经典轨道不存在了。
Born统计学说---电子衍射图的统计学解释:
Born认为具有波动性的粒子,虽然没有确定的运动轨道,但在空间任一点出现的概率与该处的波的强度成正比。
5. Schrödinger方程-微观粒子的量子力学模型
①二阶偏微分方程(不要求!)
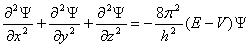
 :波函数;E:总能量;V:势能;m:质量;h:普朗克常数;x,y,z空间坐标
:波函数;E:总能量;V:势能;m:质量;h:普朗克常数;x,y,z空间坐标
②求解薛定谔方程,就是描述微粒运动状态的波函数以及与该状态相对应的能量E;
③波函数是空间坐标的函数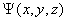
把直角坐标转化成球坐标后:
?
R(r) 径向波函数:只与半径r有关。
 角度波函数:只与角度
角度波函数:只与角度 有关,与半径r无关。
有关,与半径r无关。
为了得到合理解,波函数必须满足一定条件,因此需引入3个量子化的常数项物理量 即3个量子数:n, l, m。
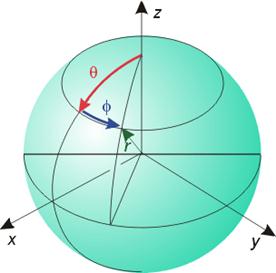 x = r sinq cosf
x = r sinq cosf
y = r sinq sinf
z = r cosq
(q=0~180°, f= 0~360°)
6. 描述电子运动状态的四个量子数
n 主量子数(principal quantum number)
物理意义:决定电子运动的能级高低和电子离核平均距离的远近。
n的取值:1,2,3,…
与n取值相对应的电子层符号:K,L,M...
l 角量子数
物理意义:
确定原子轨道(电子云)的形状;
在多电子原子中,与n一起决定电子运动的能量
l的取值范围:0,1,2,3 …,n-1
对应的电子亚层符号:s, p, d, f, ...
m 磁量子数
物理意义:决定原子轨道的空间伸展方向;
m的取值:0, ?1, ?2, …, ?l,共2l+1个值;
简并轨道-等价轨道:磁量子数与电子的能量无关;
一组特定的n, l, m值确定一条原子轨道(波函数)。
 自旋量子数(Spin quantum number)
自旋量子数(Spin quantum number)
描述电子的自旋运动:取顺时针和逆时针方向;
取值:=1/2
每条原子轨道中最多可容纳两个自旋相反的电子。
Note: n, l, m由求解薛定锷方程时所引入的。

7. 波函数
1) 波函数与原子轨道
波函数j是描述核外电子在空间运动状态的数学表达式---Schrödinger方程的解。
基于波的概念,波函数j (强度、振幅)没有明显的物理意义,但电子在某一时间(t),某一点(x,y,z)的波函数的平方的绝对值|j|2却有明显的物理意义。
|j|2与核外某处出现电子的概率成正比。因此|j|2代表单位体积内发现一个电子的几率,即称为概率密度。
m 磁量子数 (magnetic quantum number)
物理意义:决定原子轨道的空间伸展方向;
m的取值:0, ±1, ±2, …, ±l,共2l+1个值;
简并轨道(degenerate orbital)-等价轨道:磁量子数与电子的能量无关;
一组特定的n, l, m值确定一条原子轨道(波函数)。
ms 自旋量子数(Spin quantum number)
描述电子的自旋运动:取顺时针和逆时针方向;
取值:ms = ± 1/2
每条原子轨道中最多可容纳两个自旋相反的电子。
Note: n, l, m由求解薛定锷方程时所引入的。跃迁规则:电子可在不同轨道间跃迁,
结论:对于氢原子,计算值与实际值惊人的吻合,误差值小于0.1%,波尔成功地解释了氢原子光谱,上升为波尔理论。
原子轨道(orbital) ? 波函数?n, l, m
一个电子的可能空间运动状态(可用一个波函数?n, l, m表示),称为一条原子轨道。
波函数与原子轨道可以认为是同义词。
波函数描述的原子轨道与Bohr假设原子轨道本质不同。
2) 波函数的图形描述-波函数角度分布图和径向分布图
波函数角度分布图:角度波函数Yl, m(q, f)随角度q, f作图。
角度分布图与n无关,只与l, m有关。
只要l, m相同,则角度分布图完全相同。 例:2px,3px,4px
l相同,m不同时,形状(轮廓)一样,但伸展方向不同。例:2px, 2py, 2pz
波函数角度分布图有正负之分。如:p38图
波函数径向分布图:Rn, l(r)–r 径向波函数R(r)对r作图。
3) 电子云的图形表示-电子云径向分布图和角度分布图
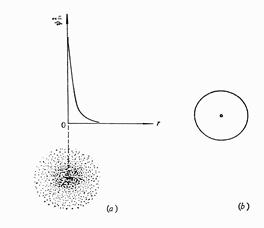
波函数的物理意义: |Ψ|2 原子核外出现电子的概率密度。
电子云图:以黑点的疏密表示电子在核外某处出现的概率密度分布的图形。
物理意义:电子出现的概率密度 Û 电子云浓度
Note:电子云是电子在核外空间出现概率密度分布的形象化描述。 电子云中的点不代表电子,而是几率密度的形象化表示。
电子云常用小黑点的疏密程度表示电子出现的概率密度。
电子云界面图:为表示电子云在空间分布的形状可将电子云密度相同的点连接起来,就围成一个曲面称为等密度面(或等值面)。通常把电子出现概率大于90%都包括在内的等值面做为电子云的界面来表示电子云的形状,这样的图叫做电子云的界面图。


 电子云
电子云
电子云角度分布图
由电子云角度分布函数Y2l, m(q, f)对(q, f)作图而得;
电子云角度分布图的形状,见p34图1-5
电子云角度分布图与波函数角度分布图的区别、联系:
Y2l, m(q, f)表示电子在空间(q, f)方向上出现的概率密度, 无正负之分。
Yl, m(q, f)反应了电子在空间(q, f)变化时的空间分布情况,亦称原子轨道角度分布图。具有方向性,有正负之分。
Yl, m(q, f) Y2l, m(q, f)
正,负 正
胖 瘦(Y2l, m(q, f) £1)
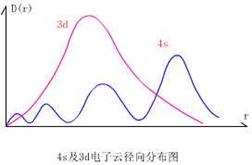
电子云径向分布图
电子云径向分布函数 :D(r) = |j |2·4pr2 = R2n, l(r) ·4pr2 (*概率密度 × 体积 = 概率)
物理意义:离核 r 处“无限薄球壳”里电子出现的概率,D值越大表示电子在该球壳里出现的概率越大;
R2n, l(r)与D(r)的区别联系: 概率密度与概率的关系。R2n,l(r)电子云径向密度分布,电子在核外出现的概率密度,与半径成反比。
电子云径向分布(函数)图--- D(r)-r作图,见p39图1-8
表示电子出现的概率随半径r的变化。指电子在核外距离为r的一薄层球壳中出现的概率随半径 r 变化时的分布情况。
因为球壳体积4pr2·dr 与半径 r 成正比,而R2n, l(r) 为概率密度与半径 r 成反比。 故电子云径向分布图会出现峰值。
出现峰值个数规律:n-l。

2s, 2p 电子云径向分布函数(r2R2 n, l( r))
出现峰值个数规律n-l

小结2:波函数(y)和电子云(y2)的空间图象
波函数:径向函数 × 角度函数
y n, l, m (r, q, f) = R n, l (r) ×Y l, m (q, f)
R n, l (r) : 波函数的径向部分,由n, l决定
Y l, m (q, f): 波函数的角度部分,由l, ms决定
电子云 Û 概率密度 y2n, l, m(r, q, f)=R2n, l(r)·Y2l, m(q, f)
R n, l (r) – r 波函数(y )径向分布
R2 n, l (r) – r 电子云 (y 2)径向密度分布
D(r) – r即r2R2 n, l (r) – r电子云 (y 2)径向(概率)分布 (D(r) = |j |2·4pr2 = R2n, l(r) ·4pr2 (电子在离核半径为r单位厚度的薄球壳内出现的概率:概率密度 × 体积 = 概率。)
Y l, m (q, f) 波函数(y )角度分布(+, -)
Y2 l, m (q, f) 电子云(y 2)角度(概率)分布
小结3:四个量子数和电子运动状态

l = 0, 1, 2, ……, (n-1); m = 0, ±1, ±2, ……, ±l
三、基态原子电子组态——核外电子排布规律与填充
1. 多电子原子轨道似能级图
单电子体系:电子能量只与主量子数n有关;
多电子体系:电子能量与n、l有关。
能级概念:不同运动状态的电子(或原子轨道)具有不同的能量,有高低之分,故称之为能级。
鲍林近似能级图---只适用于多电子原子*
近似能级图反映了原子轨道能量的相对高低,电子填充时按此顺序进行,故又称电子填充顺序图;
各电子层能量随n依次增大。
当l相同时, 轨道能级由n决定。Ex: E2s
当n相同时,各亚层轨道能级由l决定。Ex:E4s
当n, l不同时,n>3时则出现能级交错现象。Ex:Ens < E(n-1)d (n>3) —钻穿效应
能级分裂 :
n 同,l 不同,
如:E3s < E3p < E3d
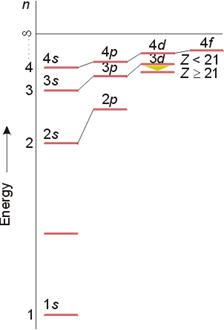
能级交错:
n, l 均不同,
E4s < E3d (Z < 21)
2. 构造原理

3. 核外电子排布规律
泡利(Pauli)不相容原理:
同一原子中,不可能出现n、l、m、ms完全相同的两个电子,即:每个原子轨道中最多只能排布两个自旋相反的电子。
能量最低原理:电子填充由低到高能级;
洪德(Hund)规则
同一亚层中,最多轨道规则。即:电子分布在角量子数 l 相同的简并轨道上时,总是尽可能分占不同的轨道,且自旋平行。
洪德规则特例:简并轨道中的电子排布,当电子处于
半充满:p3,d5,f7-最稳定状态
全满:p6,d10,f14、全空:p0,d0,f0-相对稳定状态
核外电子构型式或电子排布式
几个Hund规则特例:Cr, Cu, Mo等的电子排布式;
电子结构式中能级的书写顺序与电子填充顺序有不同;
原子失去电子的顺序与电子填充顺序相反。
四、元素周期表和元素周期系
1. 元素周期表(Periodic Table)的构造
1) 电子层结构与周期(Periods)
周期数=电子层数=最外主量子数 各周期中元素数目
特短周期-第一周期 2
短周期-第二、三周期 8、8
长周期-第四、五周期 18、18
特长周期-第六、七周期 32、32
各周期中能容纳元素最大数目为
2) 电子层结构与族(Family or Group)
价电子、价电子层、价电子层结构的概念
周期表共分8类16族---I~VIII 8类,A(主)、 B(副)两族
主族:族数=价电子数;
副族:除VIII、IB、IIB族外,其它的族数=价电子数。
3) 电子层结构与区(Blocks)
元素周期表共分:s、p、d、ds、f 5个区
s区:ns1-2
p区:ns2np1-6
d区:(n-1)d1-10ns1-2
ds区:(n-1)d10ns1-2
f 区:(n-2)f1-14(n-1)d0-1ns2
4) 元素的分类
主族元素(main group elements)-s区、p区
过渡元素(transition elements)-d区、ds区
内过渡元素(inner transition elements)-f区 稀土元素
(4f层) 镧系元素(lanthanide)
(5f层) 锕系元素(actinide)
元素周期表构造规律
周期数 = 电子层数 (主量子数n,7个)
族数=最外层电子数 (主族,8个)=外围电子数(副族10个)
价电子构型与价电子数
S区, (ns)1-2 ; p区, (ns)2(np)x; d区, (n-1)s1-2ndx
电子排布的周期性决定了元素性质的周期性
2. 元素基本性质的周期性
1) 原子半径(atomic radius)-数值只有近似的意义
共价半径(covalent radius)-共价键
金属半径(metallic radius)-金属键
范得华半径(van der waals radius)-分子作用力
*三种半径相差很大,一定要选择同一类型的才能比较。
原子半径的周期性
同一周期自左向右原子半径逐渐减小。同一周期,主族元素的原子半径减小幅度较大,而副族元素的则较小。
原因:电子层相同,从左到右,有效核电荷数逐渐递增。
同一族自上而下原子半径依次增大。与主族相比,同一副族元素的原子半径增大幅度较小。
第五、六周期同族元素原子半径非常接近—镧系收缩;
2) 电离能 I(ionization energy)
电离能:基态气体原子失去一个电子成为气态离子所需的最小能量称为电离能。
对于多电子原子,可以有
规律性:
同一元素,各级电离能的大小顺序为:
同周期内元素的电离能自左向右依次增大;
周期内 由左向右有起伏, 原因: hund规则特例引起的.
由左向右有起伏, 原因: hund规则特例引起的.
同族内元素的电离能由上向下逐渐减小。
主族元素原子的变化明显;
副族元素的变化幅度较小。*特例:镧系收缩。
物理意义:
衡量元素的原子气态时失电子的能力--判断元素的化学性质。通常以I1的大小判断,越小,元素金属性越强。
原子结构实验证明手段---证明原子结构的分层。
3) 电子亲合能 EA(Affliction energy)
元素的气态原子在基态时获得一个电子成为一价气态负离子所放出的能量称为电子亲和能。当负一价离子再获得电子时要克服负电荷之间的排斥力,因此要吸收能量。
电子亲合能反映气态非金属得到电子的能力!
变化规律性
同一周期:从左到右,Z* 增大,r 减小,最外层电子数依次增多,趋向于结合电子形成 8 电子结构,E 的正值增大。卤素的 A 呈现最大正值,ⅡA为负值,稀有气体的 A 为最大负值。
同一主族:从上到下,规律不很明显,大部分的 E正值变小。特例: E(N)为负值,是 p 区元素中除稀有气体外唯一的正值。 A 的最大正值不出现在 F 原子而是 Cl 原子。
4) 电负性 ? (electronegativity)
表示分子中的原子对成键电子的相对吸引能力,用 表示。
表示。
物理意义:
电负性越小,吸引电子能力越小,金属性越强,非金属性越弱。
是元素化学活泼性的衡量标准,分界值2.0;
预估计化学键的键型。一般电负性相差大于1.7为离子键。
Pauling电负性
Pauling电负性为相对值,规定H的为2.1。
一般规律:
同一周期中,元素电负性由左向右增大;
同一族中,元素电负性由上到下减小。
*周期表中:
左下角Cs最小(0.79), 最活泼金属;
右上角F最大(4.0), 最活泼非金属。
-
高中无机化学知识点归纳
无机化学知识点归纳一、常见物质的组成和结构1、常见分子(或物质)的形状及键角(1)形状:V型:H2O、H2S直线型:CO2、CS2…
-
无机化学 知识点总结
第一章物质存在的状态2一、气体...2二、液体...3①溶液与蒸汽压...3②溶液的沸点升高和凝固点的下降...3③渗透压...4…
-
20xx高考必备--高中无机化学知识点总结
高中无机化学知识点总结1熟悉元素周期表和元素周期律电子排布和周期表的关系化合价和最外层电子数元素所在的族序数的关系包括数的奇偶性微…
-
无机化学知识点归纳
无机化学知识点归纳一常见物质的组成和结构1常见分子或物质的形状及键角1形状V型H2OH2S直线型CO2CS2C2H2平面三角型BF…
-
无机化学知识点归纳
成就今天开创未来无机化学知识点归纳一常见物质的组成和结构1常见分子或物质的形状及键角1形状V型H2OH2S直线型CO2CS2C2H…
-
中级无机化学[第四章有机金属化合物] 山东大学期末考试知识点复习
第四章有机金属化合物1.有机金属化合物有机金属化合物指含有至少1个M—C键的化合物,也称为金属有机化合物。广义地,有机金属化合物包…
-
中级无机化学[第九章稀土元素] 山东大学期末考试知识点复习
第九章稀土元素1.镧系元素与稀土元素镧系元素:57~71号共15种元素,即镧(La)、铈(Ce)、镨(Pr)、钕(Nd)、钷(Pm…
-
无机化学知识点归纳
无机化学知识点归纳一常见物质的组成和结构1常见分子或物质的形状及键角1形状V型H2OH2S直线型CO2CS2C2H2平面三角型BF…
-
20xx高考必备--高中无机化学知识点总结
高中无机化学知识点总结1熟悉元素周期表和元素周期律电子排布和周期表的关系化合价和最外层电子数元素所在的族序数的关系包括数的奇偶性微…
-
无机化学知识点归纳
成就今天开创未来无机化学知识点归纳一常见物质的组成和结构1常见分子或物质的形状及键角1形状V型H2OH2S直线型CO2CS2C2H…
-
高中化学无机推断知识点总结
一、有机物结构与性质梳理1、物理性质(1)、状态固态——饱和高级脂肪酸、脂肪、TNT、萘、蒽、葡萄糖、果糖、麦芽糖、淀粉、纤维素、…