����ɫ�ײ���
ʵ��������ɫ�ײ���
һ��ʵ��Ŀ��
1��ѧϰ ����ɫ����ԭ���ͷ�����
2��ѧ�����ñ���ɫ�����ᴿ�л�������Ĺ淶������
2�����ձ���ֵ�ļ��㷽����
3���˽����ֵ��Ӱ�����ء�
����ʵ��ԭ��
�����Ļ���ԭ�������еIJ���ϵͳ���ɻ������ܵ�������ɣ�һ���ǹ̶��࣬��һ���������ࡣ���û�����и����������ѧ�����ϵIJ��죨����������������״�ʹ�С�����Ӽ��ԡ���������������ϵ������ָ��һ���¶��´ﵽƽ���������������Ũ�ȵı�ֵ��һ���������ȣ���ʹ������Բ�ͬ�̶ȷֲ��������У������Բ�ͬ���ٶ��ƶ������ձ˴˷ֿ���
�������ࣺ��ɫ�ס�ֽɫ�ס�����ɫ�ס�����ɫ�ס� Һ��ɫ��
����ɫ��(thin layer chromatography)����TLC��ʾ���ֳƱ�����������ڹ̣�Һ����ɫ�ס����������õı���������ʺ���������ѧԭ���IJ�ͬ���ɷ�Ϊ��������ɫ�ס����䱡��ɫ�ס����ӽ�������ɫ�����象��ɫ�ȡ�
��������ɫ�ײ��ù轺�����������������̳ɱ��㣬����Ʒ��ëϸ�ܵ���ԭ�㴦�����ƶ���չ���������ʽ�����������������������չ������ǰ�ƶ��������µ��������������ֻᱻ�������µ���չ�����ֻὫ�������������εĽ���-����-�����Ĺ��̣����ʾͻ�����չ�����ƶ���������ǿ��������չ�����ƶ���������������������չ�����ƶ��죬������ͬ������ڱ�����Ͼ͵��Է��롣
һ�������������������ƶ��ľ�����չ���������������ƶ��ľ���ı�ֵ��Ϊ�û��������ֵRf



������Ҫ�Լ��Ͳ���
�죨A������ͪ��B�������ȩ��C���������D������������轺G���������壻�����飺����������5��1��6��1����0.5%�ȼ���ά��(CMC-Na)ˮ��Һ��ëϸ��(�ھ�С��1mm)
�ġ�ʵ��װ��
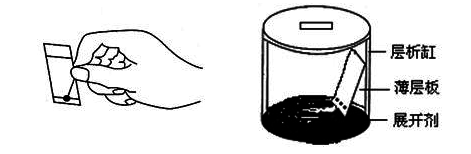
����ɫ��ʾ��ͼ
�塢ʵ�鲽��
�ư壨����ƽ�̷���
��1������7.5cm×2.5cm��Ƭ��ϴ������ˮ��
ȡ7.5cm×2.5cm�ز�Ƭ2�飬��ȥ�۷۲�ϴ������ˮ��ϴ����������ˮ�Ҵ��У�ȡ�����ɡ�ȡ��ʱ��ָֻ�ɽӴ��ز�Ƭ�ı�Ե�����ܽӴ��ز�Ƭ���档
��2��������3g�轺G��8mL0.5%�ȼ���ά����ˮ��Һ��С�ձ��н��ȡ�
��50 mL�ձ��У�����Լ3g�轺������0.5%�ȼ���ά����ˮ��Һ8mL�����ɺ�״��
��3���̲㣺��ţ�dz��˺�״���㵹����������Ƭ�ϣ���ʳָ��Ĵָ��ס����Ƭ����ǰ��������ҡ�ڶ����������Σ�ʹ�����ĺ�״����ȵ������ز�Ƭ�ϡ�ÿ���̶��顣
��4���������Ϳ�ù轺G�ı���������ˮƽ�ij�����Ƭ�ϣ����·���0.5 h��������䣬����������110 �棬����0.5 h��ȡ���������������б��á�
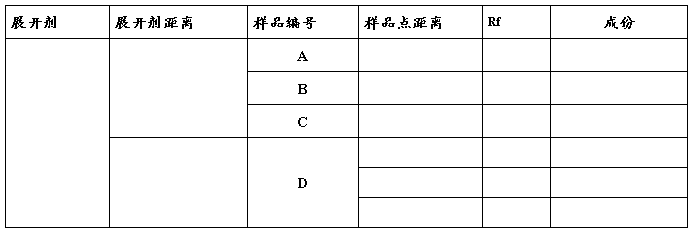
������
��1�����ߡ�����ʼ�ߺ�ǰ����
������2��ëϸ�ܵ��������ߵ��С�Ͱߵ���ࡣ���ھ�С��1mm��ëϸ��ȡ��Ʒ��Һ���ھ��뱡����8~10mm������ֱ������Ӵ�����壬�ߵ�ֱ��ҪС��2mm��һ�鱡���ɵ�2����Ʒ��ע�Ᵽ��һ���ľ��룬���ߵ㲻��̫���ߡ�
չ����չ����ѡ��չ������������б���з���ȡһ�иǵĹ��ƿ��ɫ����������չ�������û�����U��������=3��5�U1����չ�����߶Ȳ�Ҫ����5mm��������û�ߵ㣬Ȼ���ѵ����Ʒ�ı�������ɫ�����У��ǽ�����չ�����������ӽ����������ʱ�����ӣ�Ѹ����Ǧ�ʻ�С����ǰ����һ�Ǻ�ȡ�������ɡ�
��ɫ��ֱ�ӹ۲첢��ȡa��bֵ���������ֵ���������۹۲����ɼ��İߵ㣬Ȼ����������߷������¹۲�ӫ��ߵ㣬����С�����Ṵ���ߵ��������������ʢ�е�Ƭ��ƿ�н�����ɫ��
����ʵ����
�ڶ�ƪ��TLC(�������ɫ��)����ԭ����Ӧ��
TLC���������ɫ�ף�����ԭ����Ӧ��
һ�����������TLC�����
��������ǽ�����������֧�ּ�����ʱ����̻��������ȵ�����һ�鲣���ϣ��γɱ��㡣�����������Ʒ���ڱ����ϣ�Ȼ�������˵��ܼ�չ����ʹ�������Է���ķ��������ڲ����ڱ����Ͻ��йʶ�������
���������һ���������ٵIJ����������������������ڴ����ʵļ�����Ҳ�����ڻ����ķ��롢�ᴿ�������IJⶨ��������ͨ�����������������ȷ��������ʱ��ϴ�������� ���ݷ����ԭ����ͬ������������Է�Ϊ���ࣺ���������̳ɵı��������еIJ���Ϊ����������������������г��õ�������Ϊ�������轺������ά�طۡ��轺��������Ϊ֧�ּ��̳ɵı��㣬���ڷ��䱡�������
����TLC���̶���Ϊ�����������������轺�����϶��ã�
TLC��
����TLC���̶���ΪҺ̬��ͨ��Ϊˮ�����̶���������֧�ּ��ϡ�
��һ����������Ļ���ԭ����
����������Ҫ����������������Ʒ�и��ɷ�����������ͬ����չ���������ǵĽ����������IJ�ͬ��ʹ���ɷִﵽ���롣
����������Ҫ��������������������������ϡ��������������»����Ȳ���������ǿ�Ⱦ��������������������������ܱ������ɷֵ�����Ӱ�죬����չ�����������йء�
1.��������������ڹ轺������ϣ���Ʒ�е����ɷ������ֽṹ���Ƶ�Ⱦ�ϣ���չ�������Ȼ�̼�������¡���չ�����ͱ����֮�䲻�ϵز������������������������ٽ��������������ڶ���ż�����ļ��Ա�ż�����ļ�����ǿһЩ�������Ľ��������ż�����ܵ�������������ǿ��ż�������Ӷ������߷��롣
չ�������Ժ��ڱ�������γ������ߵ㣬������еijɷֵ��Է��롣
��ҩ�е���Ч�ɷָ��Ӷ������ṹ�����߲��١��ر��Ƕ�δ֪�ṹ�ijɷַ�������Ʋ������������IJ�����������Ҫ����ֻ������Ƴ����õIJ����������پ������Ľ����ſ��ܶ�δ֪������֪�ɷֽ��гɹ��ط��롣Ȼ�����̸���Ͻ�һ���ķ����о���
Ҫ��Ƴ�������Ч�IJ���������������Ϥ�������������ѡ��Ļ���Ҫ�졣����Ա������������ѡ����һ�������ܡ�
�������������������������ǣ���Ʒ��֡���������չ��������������������ʵ������Э���������ߵĹ�ϵ��Ѱ��һ���ܹ���Ч������Ʒ��ֵ���Ϸ�����ʵ�ʲ���Ҫ�졣�����ԣ�һ�в���������Է�������Ҳ����Χ�ƴ��������Ʒ��������е�����Ӧ�ġ��ɼ�������������ѡ��������Ʒ�еĸ����Ϊ���ģ��ʵ�����չ�������������ļ�����ʵ�ֵġ�
�������������������ѡ��
1.������
��������ѡ��������õ�����������ѡ��ԭ�����������ͬ����Ҫ�������ڱ������Ҫ����������֧�ּ��������ȸ�ϸ��һ��ӦС��250Ŀ����Ҫ�����Ⱦ��ȡ����ڱ����������������Ԥ�Ʊ���һ���Ȳ��˹��ߣ��Ԣ�Ϊ�ˡ���չ���������污������ȴ�ϸ��������������Խϸ��չ��������Ӧ���̣�һ�㲻����10���ף����������ɫ����ɢӰ�����Ч����
չ������ѡ��������������������Ϊһ��ֵʱ����������Զ���ֵ���Ʒ�ܷ�������ķ��룬������չ������ѡ���в�ҩ��ѧ�ɷ���֬���Գɷ��У����¿ɰ��伫�Բ�ͬ����Ϊ���ԡ������ԡ��м�����ǿ���ԡ�
����˼·�Ǹ��ݴ�������Ʒ��ֵļ�����ȷ���������ļ��ԣ����͡���ȣ���չ�����ļ��ԣ���Ҫ�ܼ��������ܼ��������ܼ��ȣ���ҪЭ������������������չ��������������ɷ�����֮��Ĺ�ϵ�����ܵõ�����ı�����������
2.��������
���������Ļ���Ҫ���ǿ���ϸ��(��������õ�����������ѡ��ԭ�����������ͬ����Ҫ�������ڱ������Ҫ��������/֧�ּ������ȸ�ϸ��һ��ӦС��250Ŀ����Ҫ�����Ⱦ���)����
С���ȣ��༴�нϴ�ı�������ʵ����������ԣ�����������Ʒ��֡���Ʒ�ܼ���չ����������ѧ��Ӧ�����������ܼ���չ���������ܽ⡣��ע��ʵ�ʲ����У���ͨ��ѡ��ͬ�Ļ�¶ȶ����������Խ��е��ڣ������ڱ����������������Ԥ�Ʊ���һ���Ȳ��˹��ߣ��Ԣ�Ϊ�ˡ���չ���������污������ȴ�ϸ��������������Խϸ��չ��������Ӧ���̣�һ�㲻����10���ף����������ɫ����ɢӰ�����Ч����
��õ��������ǣߣߣߺͣߣߣߣߡ����⣬�г��ϻ�������þ������þ��̼��þ��������������̿�ȡ����У�ֻ�л���̿�ǷǼ�����������������Ǽ��������������Ƕ�ˮ�ͼ��Դ�Ļ�������������ǿ��
��1�� ��������
��ѧʽΪAl2O3�������������������м��ԡ����ԡ��������֣�������������Ӧ�ýϹ㡣 ���������ص㣺
A: ������Ϊ��������ǿ�ļ����������������������Ի����Ե���֬�Ի�����ķ��롣ͨ���������������������������ĺ�ˮ���йء���ˮԽ�࣬��������ԽС����������ԽС�������������京ˮ�����ٽ�����Ի���Ϊ�弶��
B: �������������������������ĺ�ˮ���й�
��2�� �轺��
����ʽΪSiO2?XH2O�������ù轺��һ�ֶ�������ʣ����Ĺ����������ṹ�������ܲ����Թ贼������Si��OH�������ּ��ԵĹ贼���ܺ���������γ���������������� �ص㣺
A���轺����������������������������������Ҳ�뺬ˮ���ʸ�����ء�����л��ᡢ�ӷ��͡����ࡢ���ա���ͪ��������������ȳɷֵķ������ã��������������ȼ������ʵķ��롣
B���贼���Խ��������ԣ�������轺ֻ���������ԡ������Գɷֵķ��룬���Գɷֲ����������롣
���ڽṹ�ľ������ã��������Ļ�¶ȿ��Ժܸߣ�150�棭160�棩�����轺�Ļ�¶�ȴ����̫�ߣ�105�棭110�棩��һ������500�棬�贼�������ˮ��ʧ�
C���轺�Ļ�¶�ͨ��Ϊ105�棭110�棬���ܹ��ߡ�
������Ʒ������Ժͼ��Դ�Сȷ�����������Ժ���ȷѡ��չ����Ҳ�DZ�֤�����Ϻõķ������Ҫ������
3. չ������
չ�����������������������Ʒչ�����ܼ���
չ�����ü����ʵ����ܼ������Ѿ�������Ʒ�ı����һ�ˣ�ƾ��ëϸ���ô�����Ʒ�ڱ�������ƶ�������ʹ��Ʒ����IJ������̡�
չ�������������ֻ����������ϵ��ܼ��һ���ı�����ɵ��ܼ�ϵͳ������ܼ�ϵͳ��չ�����õ��ܼ�����չ�����������ܼ�ϵͳ�������ܼ�������չ��������չ�����̡�
4. ������ɷ֣�
������ɷ֣�����������Ҫ��ɷ������Ʒ�е���Ч�ɷ֡�
������ɷֵļ��Ծ�������ĸ�˽ṹ���ͼ������ż��ԡ�������������Ժ�չ�������Թ̶�����������£�������ɷֵļ���Խ����������������Խǿ��չ������Խ�̣�������ɷּ���Խ������������������Խ��չ������Խ��
һЩ���ŵļ�����Դ�С˳���Ѿ��ڿα�P31�г���
��Ȼ���������ʵļ��Դ�С�ж�ʱ����Ҫ��Ϸ��ӽṹ�ľ�����������жϡ�
��֮��ѡ���������ʱ����������Ʒ�жԱ�����ɷֵļ��ԣ������жϻ�ȷ������������ͻ��ԣ���̽������չ������
��ѡ���������ʱ�����������Ʒ��չ�������������������������Ͽ��ǡ�
��ʷ�����������������磬���ɶ�������ѧ�Ҵ�ά����̼�����Ϊ�̶��࣬ʯ������Ϊ������������ֲ��ɫ�صġ�������������֮�⣬��һ����IJ���������Һ����Ϊ�̶����Һ���
����Һ��̶��ࣨ��Ϊˮ���������ڹ�̬���������ϣ���Щ�������ʶ���һ�ֶ�������ʡ���ЩҺ̬�̶��౻����̶������У��ͳ�ΪҺ��Һ������������
���������䱡�������
�ü����ܼ������ڹ���֧�ּ������γɵĻ����̳ɱ��㣨��װ������Ȼ�����������������������ü��Խ�����չ��������ϴ�Ѽ�������չ����
���������һ��ԭ������չ�������У����ɷ��ڹ̶����������֮�����������ϵķ��䣬���ڸ��ɷ��������ķ���ϵ����ͬ��������Դﵽ������Ŀ�ġ�
��CS��ʾij�ɷ��ڹ̶����е�Ũ�ȣ�Cm��ʾij�ɷ��ڹ̶����е�Ũ�ȡ��� ����ϵ����K��CS/Cm
����Ӧ�þ߱��Ļ������������ԡ����ĩ�����������ԡ���ϴ�Ѽ�֮�в��ܽ⣻���ܹ����չ̶������������ܴ����屾��������50%���ϣ���ʹ���������ɵ�ͨ�����������յĹ̶��࣬���Ҳ��ı��ܼ�ϵͳ����ɡ��ܹ�������Щ�����������ǹ轺������������ά�ط۵ȡ�
����������õĹ̶���һ��Ϊˮ������ˮ��Һ���ᡢ����뻺��Һ�������������ͼ�������ˮ�ԣ��ȡ�
����������õ�������ѡ����ˮ���ܣ����ܣ����л��ܼ�����ʯ���ѡ�����±�����ࡢ֬�ࡢͪ�ࣨ�綡ͪ�������ࣨ�������촼���Ȼ������ǵĻ���
��������Ի�����и��ɷֵķ��룬��Ҫ�����ڸ��ɷַ���ϵ���IJ��죬һ��˵������������ɷ־������ã��ر���������ˮ���ԡ���ˮ�����ʶ������������л��ܼ����ߡ�������ǡ���ֲ���һ������������������������������������֬�����ʣ��IJ��㡣���ǣ����ڷ�����������Ʒ���������٣���ˣ������������������������ʱ��������ʹ����������轺������������
һ��������������������PC����������Ӧ���ڷ�����������
���ģ� ������Ʊ���
1������Ĺ����ɣ�5��15�M��5��20�M��10��10�M��20��20�M��С�IJ�������߲���ְ塢���ϰ塣
2�������ķ��ࣺ���壨�ư�ʱ����ճ�ϼ�����Ӳ�壨�ư�ʱ����ճ�ϼ�����
3��������Ʊ������P32��
4��Ӳ����Ʊ�����ϸ��ʾ�������ֹ��̰壬�˽ⱡ��Ϳ������ʹ�÷�����
���壩 ��������IJ������裺
1��������
A: ��Ʒ���ܽ⣺����Ʒ����չ��������������ӷ��Ըߵ��л��ܼ���
B: �����������ëϸ�ܣ�0.5mm���£�����רҵ���������е�����
������Ҫ������λ��Ӧ�ھ���ױ�1-1.5cm�����������ʵ�������ֱ��С��2-3mm����������������������ʱ�����Ϊ2cm�Ҵ���ͬһ��ֱ���ϡ�
����������������PC��TLC���ϲ�ͬ������Ʒ����չ������ɫ��۲����������Դ�ȷ�������Ʒ������
2��չ����
�������ϵ��ܼ���ֻӸɺ�����������ܱ������У�ʹ�ʵ���չ�����ӱ����һ������һ�˽��н���չ���Ĺ��̡�
Ҫ���ܱ�������ѡ�ò����ס��걾�ס��걾Ͳ�ȣ�չ����ʽ�����з������з�֮�֣�չ�������е���˫���չ���ȡ����P33��
ע����������ձ��͡�����Һչ�������㲻������չ�����У��������ʱ������б���롣
3����ɫ��
���ʵ��ķ��������ʵ�����ɫ���������㣬ʹ���Ͽ����Ѿ�����ĸ��ɷְߵ���ʾ�������Է����������ߵ�ı���ֵ���Ӷ��ṩ�����붨�������ݡ�
��ɫ�Ļ��������ǣ�һ�������ա����⡢���ԣ�ӫ�ⱳ��Ҳ���������P34��
4������ֵ�ļ����붨�ԡ�������
չ����������������ɫ��������Ʒ�и����ɷֵİߵ���ܳ����˲�ͬ�̶ȵķ��룬Ϊ�˱�����ɷֵ����λ�ã����ԣ�ͨ���Ա���ֵ��Ϊ�����ߵ�λ�õ�ָ�ꡣ����ֵ�ķ���ΪRf��
Rf��(�ߵ�������ԭʼ����֮��ľ���)/(�ܼ�ǰ����ԭʼ����֮��ľ���)
ע��������������Rf ֵ�ܶ�������Ӱ�죬��ʹ�ϸ���ʵ��Ҫ�����ˣ�������������Խϲ��ˣ����㶨��ʱ�����Ʒһ��������жԱȷ�����
����չ������ѡ���볣���ܼ��ļ���
��һ��չ������ѡ��
�����������У��������������������ƶ����ٶ���չ�����ļ����йء�չ�����ļ���Խ�������ƶ����ٶ�Խ�죬չ�����ļ���ԽС����������ƶ��ٶ�����
�������Ҫ�õ��Ϻõ�չ��Ч��������������Ҫ�������Ʒ����ȷѡ���������������Ʒ�ļ��Խ��������о�����ȷ����ʵ���������˵���������չ�����������������Ϊһ��ֵʱ����������Զ���ֵ���Ʒ�ܷ�������ķ��룬������չ������ѡ���в�ҩ��ѧ�ɷ���֬���Գɷ��У����¿ɰ��伫�Բ�ͬ����Ϊ���ԡ������ԡ��м�����ǿ���ԡ�����ʵ�ʹ����У�������Ҫ�����ܼ��ļ��Դ�С����չ�����ļ������Ե�����
ͨ�����õ�һ�ܼ�չ�������ݱ����������ڱ����ϵķ���Ч������һ�����Ǹı�չ�����ļ��ԡ�Rfֵ��ѷ�Χ��0.3��0.5��Χ�����÷�Χ��0.2��0.9�����Rf�ϴ���������뼫�Խ�С���ܼ����Խ���չ�������ԡ���֮�����뼫�Դ���ܼ���
������չ�����ļ���
����չ�����ļ��Դ�������ܼ����ܼ�ǿ�Ȳ�����0���������ܼ��Ħ�0Խ����Խǿ��ϴ������Խǿ����֮Խ���������ܼ��ļ������£�
�ܼ� �е� �ܼ�ǿ�Ȳ�����0 �ܼ�ǿ�Ȳ���P` ѡ�������
������ 99 0.01 0.1
������ 69 0.01 0.1
�嶡������ 56 0.35 2.5 1
�� 81 0.32 2.7 7
���� 2.8 1
���ȼ��� 40 0.42 3.1 5
������ 97 0.82 4.0 2
����� 66 0.82 4.0 3
�������� 77 0.58 4.4 6a
�ȷ� 61 0.40 4.1 8
�������� 101 0.56 4.8 6a
��ͪ 0.66 5.1 6a
�Ҵ� 0.88 4.3 2
���� �� 6.0 4
���� 0.65 5.8 6b
�״� 0.95 5.1 2
ˮ �ܴ� 10.2 8
Snyder�����ܼ��ļ���p`�ֳ�ѡ���Բ�ͬ�İ��飬�����ijһ�ܼ���չ��������Ч�����á���ѡ��ͬ�����һ���ܼ���չ�����Ͳ����ɫ���������Եĸı䣬��ѡ���ܼ�ǿ����ͬ��ѡ���Բ�ͬ�����е���һ���ܼ����ܸı�ɫ���������
��������������Դ�λ������������࣬���ִ�ɫ������������2004.2��һ�档
����Ӧ��ʵ��
1��ҩ�P��Ȼ�����е�Ӧ��
���ݱ��˵ļ��걡��������飬�ο�ҩ��ȹ���ҩƷ�����й����ף���2000��ҩ��һ���ﲿ���д����ԵĶ���Ʒ�ı����ʵ����չ���������������������һЩ���������µķ����ͽ��������������Եģ�Ŀ���ǿ��١�����ѡ��չ������������˽�չ����ѡ��ĸ������ۣ���ο�����ר����
ѡ��չ������Ҫ�����ܼ����Ժ����ǵĻ����ԣ��ܼ��Ա���������ܽ��ԣ��Լ���������Ľṹ������ֻ����ҩ����ͨ��ʹ�õ��Թ轺Ϊ�̶�����������ౡ�㣬Ҳ�����ǰ�Ļ��ԡ� �г��ܼ����Բ��������������±Ƚ�չ������������:-0.2��ʯ���ѣ����࣬30~60�棩��ʯ���ѣ����࣬60~90�棩��������:0.0���ױ�:2.4�����ױ�:2.5����:2.7�����ȼ���:3.1�������:3.9��������:3.9�������:4.0���ȷ�:4.1���Ҵ�:4.3����������:4.4���״�:5.1����ͪ:5.1������:5.8������:6.0��ˮ:10.2
���ܼ������ԣ�һ�������������ԭ����Ҫע�⣬��������IJ����ܣ�������������״�����Ԫչ����������������ܼ����ܻ��ܣ�����Ҫͨ���������ܼ������͡����磺ʯ���ѡ������顢�����顢���顢������ͼ״���ˮ֮��ġ�
һ������ɫ�ף��̶���Ϊ���ԣ����������ʵļ���Խ����Ҫ���Ը����չ������ �˽ⱻ������ļ��Կ���ͨ��������ṹ��ã����ѻ�����ļ���ָ�������ʷ��ӻ�ѧ�ṹ�У�ͨ���ɽϼ��Բ��ֺͷǼ��Բ��������֡����������Ա�����Ϊ����С���֣����ż��Ի��Ų��ֵ����ӣ�����ļ��Ծ����ӣ�չ��������Ҳ�����ˡ�����Ϊ����ᡢ��κ�ᡢ�����ᡢ�����ᡢ��ԭ�ᡣ��Ӧչ�����ֱ�Ϊ�������顪���ѡ������� (5:5:0.1)�����������ᣭ�״�(30:1:3)���ȷ£��״������ᣨ9:1: 0.5����ʯ���ѣ��������������ᣨ3:6: 1�������ᶡ�������ᣭˮ(7:2.5:2.5)�������ڱ���塢����ֵ��ͬ��ԭ��չ�������ԱȽ�����Եģ����Ǿ��Եĺ��ߴ���ǰ�ߣ���
��������Ҫ�������ǣ���ͬ�������ô�����ļ��ԣ�����ʲô����������Ӧ��չ�����أ�
���·ֿ����۲�ͬ�����K����������Ӧ��չ������
��1�� ���Խ�С�Ļӷ������ʡ�
���磺��Ƭ��ʯ���� (30��60��)����������(17:3)�����ӷӣ�������������(9:1.5)����-�㸽ͪ����������������������(92:5:5)����Ƥ�ӣ������飭��������(3:1)����������ʯ���ѡ�������ͱ�Ϊ����ٷ����Ƚϴ���ܼ���ͨ�����ܽ�ͷ��뻯��������ã����ô�������Ϊ����Rf������ֵ�����ܼ���Ϊ�˼�����β֮��������������ԭ�������Ӱ�죬�ʵ��������Ӽ������л�������л��
��2�����Խ�С�IJ��ӷ������ʡ�
���磺�� -�����������飭�����������״�(6:2.5:1)�������飭��ͪ(5:2) ���ܹ���ױ�������������������(12:4:0.5)����չ���ȷ£��״�(40:1)����ȥ������ȷ£����ѣ�������(2:2:1)������أ����������������״�(15:2:0.2)���߱����Ҵ� (8:1)������ͪ��A����-��������-����(40:25:4) ���������������ȷ�-��ˮ�Ҵ�(9:1)������졢�����ȷ£��Ҵ�(9:1)���߱����ȷ£���ͪ(5:4:1)����������չ�������Աȼ��Խ�С�Ļӷ�������ϴ����ǿһЩ����Ϊ�������ʼ���С��ĸ�˴����Դ�Ļ���ͨ�������γ�������������ᡢ�ǻ����������ʣ�ĸ�˷�������С��ĸ�˽ṹ�в����ͽ������ӣ������dz��ֱ����������Ի��ŵ����ӣ���ʹ�������ӣ�չ��������Ҳ���������Χ�ڵ����ʺܶ࣬һ��չ������ٷ������ܼ����Դӻ����顪���ױ��������ױ������������ȷµ�˳���ռ���Ҫ��ѡ������ע�⣬�����������������ָ��Ҳ�Ƚ�С�����ⷶΧ�Ļ���������ã���Ϊճ�Դ�չ��������ɰߵ���ɢ�����⣬�ǻ������������Ҳ�в���������Rfֵ���ܼ����Ӵ������������״�������ͪ�����Ҵ����ӷ�������Ҳ�кܶ���ʻ����ǻ��ģ��������Ļӷ��ԾͿ������ף����Ӽ���������ǿ�����⣬ĸ����ʯ���ѡ�������ͱ��Ľṹ����С�����Ƹ���������轺��������������ܼ��У�������Ҫͨ�����չ�����ļ��ԡ�
��3�������ࡣ
�˲����գ��ȷ£��״���ˮ (65:35:10)10�����·��õ��²���Һ��������������������ˮ(4:1:5)���ϲ���Һ���ȷ£������������״���ˮ(15:40:22:10)10�����·��õ��²���Һ����ҩ�գ��ȷ£������������״�������(40:5:10:0.2)�������գ�������������ͪ�����ᣭˮ
(10:7:5:3)����Ƥ�գ������������������ᡪˮ(1:12:2.5:3)���ϲ���Һ������أ��ȷ£��״���ˮ(14:5:0.5)��«�����������������ᣭˮ(8:1:1)���������ʣ����ڴ����ǵĶ��ǻ��ṹ����Ԫ�ĽṹӰ���С��չ������ʹ�ü��Դ���л��ܼ����ȷ¡������������״�������������ˮ������ͼ����ʹ�ã�һ��������չ�������ԣ�����Ҳ�������ƹ轺�ǻ������ã�������β�����ڻ����Ժ轺�������������ƣ�ˮ�����ʹ�������ȵġ�
��4�����Դ��С�����л��ᡣ
ûʳ����ȷ£��������������� (5:4:1)����κ�ᡢ�����ᡢ�����ᡢ��ԭ�ᡢ����ԭ�ᡣ�������ʶ����DZ���ϩĸ�˵ģ�����ṹ�ļ��Ա����Ƚϴ������з��ǻ���������ţ������ж��ǻ���������յ�չ������࣬���Դ�ע�����ͨ��ָ����Ũ��85%���ҵģ�����ˮ��
��5��������
����С����������������״����������Ũ����Һ(12��6��3��3��0.6)(����������) ����������������-ˮ(7:1:2)����Ƽ�ȷ£��״���Ũ����Һ(20:5:0.5)���������������ᣭˮ(8:2:1)���ʲ���泥��������������ᣭ�����ᣭˮ(15:1:1:2)������NH2�贼�������ú�ǿ����ǿ����չ�������л��ᡢ�л���ɨβ�����ڼ��Ի����ʹ���������ߵ���ɢӰ���С����Ϊ������轺������ǿ��
���б�������������Ը���ĸ�ˡ����ţ�ѡ�����ƵĻ�����Ժ���������Ȼ������������Ż�����Ҫ����ʵ������ˡ����������ѵķ��룬��Ҫʹ�õ�����Ż������ģ��Ѿ������ڱ������۷�Χ�ˡ�
2���л��ϳ���չ������ѡ��
���л��ϳ�ʱ�߰����dz��е��£�չ������ѡ���������Ҫ��,ѡ���ʵ���չ��������Ҫ����.һ�㳣���ܼ����ռ��Դ�С�����˳�����д��Ϊ��ʯ����<����<��<����<THF<��������<��ͪ<�Ҵ�<�״�ʹ�õ�һ�ܼ����������ܴﵽ�ܺõķ���Ч��������ʹ�û���ܼ�ͨ��ʹ��һ�����Ժ͵ͼ����ܼ���ɵĻ���ܼ������Ե��ܼ������������ֶȵ����ã����õ��ܼ�����У�
Petroleumether/Ethylacetate, petroleumether/Acetone, Petroleumether/Ether,
Petroleumether/CH2Cl2, ethylacetate/MeOH, CHCl3/ethylacetate
չ�����ı���Ҫ������.һ����������б����ĸ��������ʲô����չ�����������ȳ���ʹ�ø���չ������Ȼ�ϳ��Ա�����ֱ���ҵ�һ������Ч���õ�չ������չ������ѡ���������ٶԵ�����ɷ������õ��ܽ��ԣ��ڿ�ʹ�ɷּ�ֿ����۴�����ֵ�Rf��0.2~0.8֮�䣬�����ⶨ��0.3��0.5֮�䣻�ܲ��������ֻ�������������ѧ��Ӧ���ݷе����У��Ƚ�С����չ������ְߵ�Բ�Ҽ��У�����ܼ�������������ơ�һ����˵���������ܼ���ϵ�Ļ����������������ˮ��ɣ��ٸ�����Ҫ����״����Ҵ������������������ܼ�ϵͳ�ļ��ԣ��Դﵽ�õķ���Ч�����ʺ���������ͪ������ȵķ��룻�еȼ��Ե��ܼ���ϵ���ȷº�ˮ����������ɣ��ɼ״����Ҵ������������������ڣ��ʺ����������㶹�أ��Լ�һЩ���Խϴ��ľ֬�غ�����ķ��룻ǿ�����ܼ�������������ˮ��ɣ�Ҳ���״����Ҵ������������������ڣ��ʺ��ڼ��Ժܴ������������ķ��롣
�ܶ�ʱ��,չ������ѡ��Ҫ���Լ����ϱ任չ������������ﵽ���Ч����������ʵ���У�Ϊ��ʵ��һ������������������Ч���룬���������˺ܶ��ֵ��ܼ���ϣ������ҵ�ʯ���ѡ�EtOAc��HCOOH��5.5��3.5��0.1������ܼ���һ��������ܼ����ʱ,���ø���/�ͼ��Ե������Ϊ1/3�Ļ���ܼ�,����зֿ��ļ���,�ٵ���������������������ܼ���,�ﵽ���Ч�������û�зֿ��ļ��ߵ�ϡ��ϡ���������ǻ��ܼ��������ڹ轺�����������������ֽ�����ʣ���չ������������һ������Ұ�����ˮ����वȼ����������к轺�����ԡ���ѡ�������ӵļ������ʣ������뿼�����״Ӳ�Ʒ�г�ȥ����ˮ�����ǽϺõ�ѡ������Ч���ĺû������ù轺���ܼ����������й�ϵ����ͬ���������Ĺ轺���ܺ�ˮ���Լ������Ĵ�ϸ�̶ȣ�����ǿ����ͬ���Ӷ����²�Ʒ��ij�����ҵĹ轺�з���Ч���ܺã�������һ�����ҵľͲ��С��ܼ��ĺ�ˮ�������ʺ����Է���Ч���������Ե�Ӱ�졣�¶ȣ�ʪ�ȶԷ���Ч��Ӱ��Ҳ�����ԣ���ʵ�������Ƿ�����ʱͬһչ���������������Rf��Ȼ��ͬչ������ѡ����Ҫ������Ʒ�ļ��ԡ��ܽ�Ⱥ��������Ļ��Ե������������ڽ��б������ʱ������Ӧ��֪��δ֪��ѧ�ɷֵ����ͣ��伫�ԵĴ��¹���������ȡҺ���ɫ�����������༫�Կ�֪������ij��Ʒ�ﺬ���ֻ�ѧ�ɷ��Ȱ����Բ�ͬ���·֣�Ȼ��ϸ�֣����ڷ���δ֪�Ļ�ѧ���ʣ�չ������ѡ��Ҳ��һ�������Ĺ��̣���Ӧ�ý�����չ�������ǣ��������ۺϺ�����
�ܼ��������������ܼ���ѡ����ַ����ϵ������������ʱ���õ��ܼ�����һ�������ܼ���ϰ���ϳ�ϴ�Ѽ������ڱ����ֽ����ʱ����չ������ϴ�Ѽ���ѡ������ݱ�������������ѡ�õ����������������߽���������Կ������ü������������в���ʱ��������������Ϊ���������ʣ�һ��ѡ���������ܼ�Ϊϴ�Ѽ�������������Ϊǿ���Գɷ֣�����ѡ�ü����ܼ�Ϊϴ�Ѽ��������ijһ���������������Խ����������������Թ�������ʯ�۴���轺������ϴ�Ѽ��ļ���������Ӧ���͡�
���������ʱ����������Ʒ�ڼ���ʱ�ɲ����ڷ������ѡһ���˵��ܼ�����Ʒ�ܽ����롣�ܽ���Ʒ���ܼ�Ӧѡ���Խ�С�ģ��Ա㱻����ijɷֿ��Ա�������Ȼ�������ܼ��ļ��ԡ����ּ��Ե�������һ��ʮ�ֻ����Ĺ��̣���Ϊ���ݶ�ϴ�ѡ���ʹ�����ڲ������ϵĸ����ɷ������ϴ�ѡ������������������ݶ�̫���Ͳ��ܻ������ķ��롣�ܼ���ϴ����������ʱ�������ܼ��Ľ�糣�����ţ�����ʾ����糣���ߣ�ϴ�������ʹ����ϵ�ϴ��˳��������ڼ�������������轺�����������ԷǼ����������������̿��������������˳���෴����ˮ����ˮס�ܼ������γɵ��������ã�����֬�����ܼ���Ϊǿ��
3�����������ʵ����ʣ����������������������ϴ�Ѽ���ͬ�������������е�����Ҫ�أ��˴˽�����������ָ������������ϴ�Ѽ��������£������ɷֵķ��������ֱ���뱻�������ʵĽṹ�������йء��Լ������������ԣ��ɷֵļ��Դ�����סǿ��
��Ȼ���в�ҩ�ɷֵ�������ӹ�����Ҫ�ģ����缫�Ի��ŵ���Ŀ���࣬��������ס�ܾͻ����Щ����ͬϵ����̼ԭ����Ŀ��Щ��������Ҳ��ǿЩ����֮��ֻҪ�����ɷ��ڽṹ�ϴ��ڲ�𣬾��п��ܷ��룬�ؼ�����������ѡ��Ҫ���ݱ��������ʵ����ʣ�������������ǿ�ȣ����ܼ������������ߵ����ϵ�����ǡ�����Ҫ���DZ��������ʵļ��ԡ��类�������ʼ��Ժ�СΪ����������ϩ�����京�����Ǽ��Ի��ţ�����ѡ�������Խ�ǿ���������������������ܼ���ʯ���ѻ���ϴ�ѡ���������ҩ�ɷֵļ��Խϴ�����Ҫѡ���������ܽ�������������һ������������õ�ϴ�Ѽ�����Ӧ��С����ijһ�ݶȵ��������Ӧ�ñ���������жϱ���������ij���ܼ�ϵͳ�еķ�����������⣬�ܷ�������ķ��룬����ѡ����ܼ��ݶ��кܴ��ϵ������ʵ��˵��������������������ϴ�Ѽ�����Ʒ����֮��Ĺ�ϵ�����ж���ֵĻ�����ֲ����֬ϵ��������ϩ����Ҩ�����ࡢ���������֬�������ݡ����Թ轺Ϊ������ʱ��ʹ��֬��������ѡ��һϵ�л���ܼ�����ϴ�ѣ���֬�и���һ�ɷּ��ɰ�
�伫�Դ�С�IJ�ͬ���α�ϴ�ѡ�
�������C-27������߰Ԫ��ɷ֣�������������ǻ���Ŀ�Ķ��ٶ���÷��룺�������߰Ԫ���ں���5���ȷµı��У��������������������ϣ��������µ��ܼ������ݶ�ϴ�ѡ�����������Խ����Ĺ���þ����������������ڹ���þ�������Խ�����ϴ�Ѽ��ļ�������Ӧ���ͣ��༴���ñ���5���ȷµı������ɽ�һԪ�ǻ���߰Ԫ����������ϴ����������һ����˵����ͬ�����в�ҩ�ɷ��ڲ�ͬ���������в���ʱ�����ò�ͬ���ܼ����ܴﵽ��ͬ�ķ���Ч�����Ӷ�˵�����������ܼ���������ɷ����ߵ����ϵ��
3������ɫ�����в�ҩ�е�Ӧ��
���������һ�ּ�㡢���١����IJ���������һ�㽫�������õ�������������ƽ���粣��Ƭ�ϣ��γ�һ������в���ʱһ���Ʊ����������ԭ�����������������ơ�
��1������������ص㣺���������Ӧ�������������ص����������ıȽϡ�
��2����������ѡ��������õ�����������ѡ��ԭ�����������ͬ����Ҫ�������ڱ������Ҫ����������֧�ּ��������ȸ�ϸ��һ��ӦС��250Ŀ����Ҫ�����Ⱦ��ȡ����ڱ����������������Ԥ�Ʊ���һ���Ȳ��˹��ߣ��Ԣ�Ϊ�ˡ���չ���������污������ȴ�ϸ��������������Խϸ��չ��������Ӧ���̣�һ�㲻����10���ף����������ɫ����ɢӰ�����Ч����
��3��չ������ѡ��������������������Ϊһ��ֵʱ����������Զ���ֵ���Ʒ�ܷ�������ķ��룬������չ������ѡ���в�ҩ��ѧ�ɷ���֬���Գɷ��У����¿ɰ��伫�Բ�ͬ����Ϊ���ԡ������ԡ��м�����ǿ���ԡ�����ʵ�ʹ����У�������Ҫ�����ܼ��ļ��Դ�С����չ�����ļ������Ե�����
��4�����ⱡ�㣺���ijЩ��������Ļ�����ķ�����������ʱ�����һЩ���ⱡ�㡣
�� ӫ�ⱡ�㣺��Щ�����ﱾ����ɫ�����������Ҳ����ӫ�⣬�����ʵ�����ɫ��ʱ��������������м���ӫ�������Ƴ�ӫ�ⱡ����в�����չ�����������������䣬����屾����ӫ�⣬����Ʒ�ߵ㴦����ӫ�⣬���ɼ����Ʒ�IJ���λ�á����õ�ӫ�����ʶ�Ϊ�����
һ���� 254nm����⼤�����Գ�ӫ��ģ����̼���Ĺ���п����һ��Ϊ��365nm����⼤���·���ӫ��ģ�������������п�䡣
�� ��ϱ��㣺���õ������������㣬��������̼ԭ������ȶ�����CһC˫����Ŀ���ȵ�һϵ�л�����粻���ʹ�����ȡ�����Ҫ����������CһC�������������γ����������͵�CһC��������������ϡ�����������������ϣ���̨������ڱ��ͳ̶Ȳ�ͬ����÷��롣����ʱ���ͻ�������������������Rf��ߣ���һ��˫���ĽϺ�����˫����Rfֵ�ߣ���һ�������ĽϺ�һ��˫����Rfֵ�ߡ����⣬��һ��˫����̨���У�˳ʽ������������ϽϷ�ʽ�����ڽ��С���ˣ�������������˳���칹�塣
�� �����PH���屡�㣺Ϊ�˸ı�������ԭ��������ԣ��������Ʊ���ʱ����ϡ���ϡ���Դ���ˮ���Ʊ��㡣����轺�����ԣ���ʱ�Լ��������������ķ��벻�ã��粻��չ�����β��������̱���ʱ����ϡ����Һ0.1��0��5NNa0H��Һ�Ƴɼ��Թ轺���㡣������ʺ�������Թ轺Ϊ������ʱ�����ȷ�-��ͪһ�״���8��2��1��Ϊչ����Rf��0��1�����ü��Թ轺������������ͬչ������Rfֵ����0��4���ҡ�˵����ʺ����Ϊ--��������
��5��Ӧ�ã�������������в�ҩ��ѧ�ɷֵ��о��У���ҪӦ���ڻ�ѧ�ɷֵ�Ԥ�ԡ���ѧ�ɷֵļ�����̽����������������
�ñ�������������в�ҩ��ѧ�ɷ�Ԥ�ԣ������ݸ���ɷ����ʼ���֪��������������Եؽ��С������ڱ�����չ��ɽ�һЩ���ʷ��룬ѡ���Ըߣ���ʹԤ�Խ����Ϊ�ɿ���
��6���в�ҩ�е�Ӧ��
�Ա�����������в�ҩ��ѧ�ɷּ��������Ҫ�б���Ʒ���й�������������������ܼ�չ���Ʒ�ͼ���Ʒ��Rfֵ���ߵ���״��ɫ����ȫ��ͬ�����������������ͬһ�������һ������л�ѧ��Ӧ��������һ�����������������Ժ˶ԡ�
�ñ��������̽�����������������ʵ���ҵij��淽�����ڽ����������ʱ�����ȿ���ѡ�ú�����������ϴ�Ѽ�����ϴ�ѹ����и����ɷֽ�������˳��ϴ�ѣ�ÿһϴ��Һ���Ƿ�Ϊ��һ�ɷֻ����壬�����ɱ���ķ���õ��ж�����顣ͨ�������Ԥ���룬�������˽�������Ʒ���������Ժ��������ڱ������������Ƚ�����ķ������������ɽ����������ڸ���������������Խ���������������ʵ��ı䣬ת��һ������������ϴ�ѵķ�ʽ������
�������롣���ñ����Ԥ����Ѱ�������ϴ������ʱ,�ٶ��ڱ���������õ�Rfֵһ��Ʒ�������еı����ʣ�R�������������ڱ���չ��ʱ������̶������������ܼ��������ϵ���������ʹ�����ϸ���λ���������ܼ����Dz��ȵģ�������ʼ�ߵĺ������ڱ����ǰ�ز��֡������ϸ���Ʋ���������������ɵõ��ӽ���ʵ��Rfֵ���ñ������ijһ��ֵķ��룬��Rfֵ��Χ��һ��������Ϊ0��85��Rf��0��05�����⣬�����������Ӧ�����в�ҩƷ�֡�ҩ�ļ����Ƽ���α�ļ�顢�������ƺ���Դ���飬�Կ��ƻ�ѧ��Ӧ�Ľ��̣���Ӧ����Ʒ����ļ�飬�м����������ѧҩƷ���Ƽ����ʵļ�飬�ٴ������������Լ���������ȣ�������Ч���ֶΡ�
ѡ���ʵ���չ��������Ҫ����.һ�㳣���ܼ����ռ��Դ�С�����˳�����д��Ϊ��ʯ����<����<��<����<THF<��������<��ͪ<�Ҵ�<�״�ʹ�õ�һ�ܼ����������ܴﵽ�ܺõķ���Ч��������ʹ�û���ܼ�ͨ��ʹ��һ�����Ժ͵ͼ����ܼ���ɵĻ���ܼ������Ե��ܼ������������ֶȵ����ã����õ��ܼ������
Petroleumether/Ethylacetate, petroleumether/Acetone, Petroleumether/Ether,
Petroleumether/CH2Cl2, ethylacetate/MeOH,
CHCl3/ethylacetate
չ�����ı���Ҫ������.һ����������б����ĸ��������ʲô����չ�����������ȳ���ʹ�ø���չ������Ȼ�ϳ��Ա�����ֱ���ҵ�һ������Ч���õ�չ������չ������ѡ���������ٶԵ�����ɷ������õ��ܽ��ԣ��ڿ�ʹ�ɷּ�ֿ����۴�����ֵ�Rf��0.2~0.8֮�䣬�����ⶨ��0.3��0.5֮�䣻�ܲ��������ֻ�������������ѧ��Ӧ���ݷе����У��Ƚ�С����չ������ְߵ�Բ�Ҽ��У�����ܼ�������������ơ�
һ����˵���������ܼ���ϵ�Ļ����������������ˮ��ɣ��ٸ�����Ҫ����״����Ҵ������������������ܼ�ϵͳ�ļ��ԣ��Դﵽ�õķ���Ч�����ʺ���������ͪ������ȵķ��룻�еȼ��Ե��ܼ���ϵ���ȷº�ˮ����������ɣ��ɼ״����Ҵ������������������ڣ��ʺ����������㶹�أ��Լ�һЩ���Խϴ��ľ֬�غ�����ķ��룻ǿ�����ܼ�������������ˮ��ɣ�Ҳ���״����Ҵ������������������ڣ��ʺ��ڼ��Ժܴ������������ķ��롣 �ܶ�ʱ��,չ������ѡ��Ҫ���Լ����ϱ任չ������������ﵽ���Ч����
������ʵ���У�Ϊ��ʵ��һ������������������Ч���룬���������˺ܶ��ֵ��ܼ���ϣ������ҵ�ʯ���ѡ�EtOAc��HCOOH��5.5��3.5��0.1������ܼ���һ��������ܼ����ʱ,���ø���/�ͼ��Ե������Ϊ1/3�Ļ���ܼ�,����зֿ��ļ���,�ٵ���������������������ܼ���,�ﵽ���Ч�������û�зֿ��ļ��ߵ�ϡ��ϡ���������ǻ��ܼ��������ڹ轺�����������������ֽ�����ʣ���չ������������һ������Ұ�����ˮ����वȼ����������к轺�����ԡ���ѡ�������ӵļ������ʣ������뿼�����״Ӳ�Ʒ�г�ȥ����ˮ�����ǽϺõ�ѡ������Ч���ĺû������ù轺���ܼ����������й�ϵ����ͬ���������Ĺ轺���ܺ�ˮ���Լ������Ĵ�ϸ�̶ȣ�����ǿ����ͬ���Ӷ����²�Ʒ��ij�����ҵĹ轺�з���Ч���ܺã�������һ�����ҵľͲ��С��ܼ��ĺ�ˮ�������ʺ����Է���Ч���������Ե�Ӱ�졣�¶ȣ�ʪ�ȶԷ���Ч��Ӱ��Ҳ�����ԣ���ʵ�������Ƿ�����ʱͬһչ���������������Rf��Ȼ��ͬչ������ѡ����Ҫ������Ʒ�ļ��ԡ��ܽ�Ⱥ��������Ļ��Ե������������ڽ��б������ʱ������Ӧ��֪��δ֪��ѧ�ɷֵ����ͣ��伫�ԵĴ��¹���������ȡҺ���ɫ�����������༫�Կ�֪������ij��Ʒ�ﺬ���ֻ�ѧ�ɷ��Ȱ����Բ�ͬ���·֣�Ȼ��ϸ�֣����ڷ���δ֪�Ļ�ѧ���ʣ�չ������ѡ��Ҳ��һ�������Ĺ��̣���Ӧ�ý�����չ�������ǣ��������ۺϺ�����
�ܼ��������������ܼ���ѡ����ַ����ϵ������������ʱ���õ��ܼ�����һ�������ܼ���ϰ���ϳ�ϴ�Ѽ������ڱ����ֽ����ʱ����չ������ϴ�Ѽ���ѡ������ݱ�������������ѡ�õ����������������߽���������Կ������ü������������в���ʱ��������������Ϊ���������ʣ�һ��ѡ���������ܼ�Ϊϴ�Ѽ�������������Ϊǿ���Գɷ֣�����ѡ�ü����ܼ�Ϊϴ�Ѽ��������ijһ���������������Խ����������������Թ�������ʯ�۴���轺������ϴ�Ѽ��ļ���������Ӧ���͡�
���������ʱ����������Ʒ�ڼ���ʱ�ɲ����ڷ������ѡһ���˵��ܼ�����Ʒ�ܽ����롣�ܽ���Ʒ���ܼ�Ӧѡ���Խ�С�ģ��Ա㱻����ijɷֿ��Ա�������Ȼ�������ܼ��ļ��ԡ����ּ��Ե�������һ��ʮ�ֻ����Ĺ��̣���Ϊ���ݶ�ϴ�ѡ���ʹ�����ڲ������ϵĸ����ɷ������ϴ�ѡ������������������ݶ�̫���Ͳ��ܻ������ķ��롣�ܼ���ϴ����������ʱ�������ܼ��Ľ�糣�����ţ�����ʾ����糣���ߣ�ϴ�������ʹ����ϵ�ϴ��˳��������ڼ�������������轺�����������ԷǼ����������������̿��������������˳
���෴����ˮ����ˮס�ܼ������γɵ��������ã�����֬�����ܼ���Ϊǿ��
3�����������ʵ����ʣ����������������������ϴ�Ѽ���ͬ�������������е�����Ҫ
�أ��˴˽�����������ָ������������ϴ�Ѽ��������£������ɷֵķ��������ֱ���뱻��
�����ʵĽṹ�������йء��Լ������������ԣ��ɷֵļ��Դ�����סǿ�� ��Ȼ���в�ҩ�ɷֵ�������ӹ�����Ҫ�ģ����缫�Ի��ŵ���Ŀ���࣬��������ס�ܾ�
�����Щ����ͬϵ����̼ԭ����Ŀ��Щ��������Ҳ��ǿЩ����֮��ֻҪ�����ɷ��ڽṹ�ϴ�
�ڲ�𣬾��п��ܷ��룬�ؼ�����������ѡ��Ҫ���ݱ��������ʵ����ʣ�������������ǿ
�ȣ����ܼ������������ߵ����ϵ�����ǡ�����Ҫ���DZ��������ʵļ��ԡ��类��������
���Ժ�СΪ����������ϩ�����京�����Ǽ��Ի��ţ�����ѡ�������Խ�ǿ����������������
�����ܼ���ʯ���ѻ���ϴ�ѡ���������ҩ�ɷֵļ��Խϴ�����Ҫѡ���������ܽ�����
��������һ������������õ�ϴ�Ѽ�����Ӧ��С����ijһ�ݶȵ��������Ӧ�ñ����
�����жϱ���������ij���ܼ�ϵͳ�еķ�����������⣬�ܷ�������ķ��룬����ѡ���
�ܼ��ݶ��кܴ��ϵ������ʵ��˵��������������������ϴ�Ѽ�����Ʒ����֮��Ĺ�ϵ����
�ж���ֵĻ�����ֲ����֬ϵ��������ϩ����Ҩ�����ࡢ���������֬�������ݡ�
���Թ轺Ϊ������ʱ��ʹ��֬��������ѡ��һϵ�л���ܼ�����ϴ�ѣ���֬�и���һ�ɷּ�
�ɰ��伫�Դ�С�IJ�ͬ���α�ϴ�ѡ�
�������C-27������߰Ԫ��ɷ֣�������������ǻ���Ŀ�Ķ��ٶ���÷��룺�������߰
Ԫ���ں���5���ȷµı��У��������������������ϣ��������µ��ܼ������ݶ�ϴ�ѡ����
�������Խ����Ĺ���þ����������������ڹ���þ�������Խ�����ϴ�Ѽ��ļ�������Ӧ���ͣ�
�༴���ñ���5���ȷµı������ɽ�һԪ�ǻ���߰Ԫ����������ϴ����������һ����˵����
ͬ�����в�ҩ�ɷ��ڲ�ͬ���������в���ʱ�����ò�ͬ���ܼ����ܴﵽ��ͬ�ķ���Ч������
��˵�����������ܼ���������ɷ����ߵ����ϵ��
������ɫ����
��һ��. ����
һ�������ա����⡢���ԣ�ӫ�ⱳ��Ҳ����
A: �������չ��¹۲죬������ɫ���ʵİߵ�λ��
B: ��������¹۲����ް���ӫ��ߵ�
C: ���л�������������IJ�����ɫ���ɫ�ߵ㡣
D: ӫ�ⱡ���⣬�۲�ӫ�ⱡ���г��ֵİ��ߡ�
E: ����ɫ�����������յ����ʣ���������ɫ����ɫ
��ɫ�����Էֳ������ࣺһ���Ǽ��һ���л��������ͨ����ɫ������һ���Ǹ��ݻ��������������������Ƶ�ר������ɫ����
��������ͨ����ɫ��
���᳣�õ���������Һ������-ˮ(1��1)��Һ������-�״����Ҵ�(1��1)��Һ��1.5mol��L������Һ��0.5-1.5mol��L�������Һ�����110�濾15min����ͬ�л��������Բ�ͬ��ɫ�� ��0.5������ȷ���Һ �Ժܶ�����Ի���ɫ��
������0.05�����������Һ ��ԭ�Ի������ڵ��챳�����Ի�ɫ��
�ܼ��Ը�������Լ� ��ԭ�Ի������ڵ���ɫ�������Ի�ɫ��
��ҺI��1�����������Һ����Һ��5��̼������Һ����ҺI����Һ��������Ӧ�á�
�����Ը�������Լ� ��1.6���������Ũ������Һ(�ܽ�ʱע���ֹ��ը)�������180�����15��20min��
�������ظ�����Լ� ��5���ظ����Ũ������Һ����Ҫʱ150�濾���㡣
��5���������Ҵ���Һ ���120��濾����ԭ�Ի���������ɫ�����ð���������Ϊ��ɫ��
�����軯��-���Ȼ����Լ� ��ԭ����������ɫ������2mol��L������Һ������ɫ���
��ҺI��1�����軯����Һ����Һ��2�����Ȼ�����Һ������ǰ����ҺI����Һ�������ϡ�
������. ר������ɫ��
���ڻ���������࣬���ר������ɫ��Ҳ�Ǻܶ�ģ��ֽ��ڸ����������õ���ɫ���о����£�
(1)����
������������������
����±�����ࡣ
��Һ��������O.1g����ˮlml����2-�������Ҵ�lOOml���ñ�ͪϡ����200ml���ټ�30����������1�Ρ�
�����������δ���˵�����������䣻
������ߵ�ʰ���ɫ��
��ӫ���أ���
��������������
��Һ��I��ӫ����0.1g�����Ҵ�lOOml��
��5��������Ȼ�̼��Һ��
����������(I)��Ȼ���ú������������ڣ�ӫ����ת��Ϊ����ӫ����(���)��ӫ����ʧ�����������ߵ�������ļӳɣ���ֹ������������ӫ�⣬�������������ڷۺ�ɫ�����ϳʻ�ɫ�� �������ڱ���������
������������
��Һ��2�������ڱ����������ı�ͪ���ȴ���(10��1)����Һ��
�����������������¹۲졣
�ܼ�ȩ������
�����������
��Һ��37����ȩ��ҺO.2ml����Ũ����l0ml��
(2)����
��3��5һ������������
�������ࡣ
��Һ��I��2����Ʒ�ױ���Һ��
��0.5������������Һ��
��O.002��������Һ��
����������(I)���ڿ����и����ҹ��������2min����ֽ��ͨ����Һ(��)30s����ˮϴ����ʪͨ��(��)15s���������������¹۲졣
���������
�������ࡣ
��Һ��I��1��������淋�0.2mol��L������Һ��
��N,N-����-�Ա�����������1.5g���ڼ״���ˮ������(128m1+25m1+1��5m1)���Һ�У���ǰ��(I)��(��)������ϡ�������105oC����5min��
�����ȩ������
�����������ӡ����༰���͡�
��Һ�����ȩ1g��������lOOml��
�����������120oC��������ɫ���
�ܶ������������ ��
�������ࡢ��ϩ���ʻ����������ࡣ
��Һ����Ʒ15mg�����ȷ�25ml�С�
�����������110oC����5��lOmin��
�������ɫ�����ʻ�ɫ�ߵ㡣
(3)ȩͪ��
��Ʒ�죯������
����ȩ�������
��Һ��I��0.01%Ʒ����Һ��ͨ���������ֱ����ɫ��
��0.05mol��L�Ȼ�����Һ��
��O.05mol��L������Һ��
��������I������1��1��10��ϣ���ˮϡ����l00m
���������㰷
����ȩ�ࡢͪ�ࡣ
��Һ����Ʒ���ᱥ����Һ��
��2��4-����������
����ȩ����ͪ����ͪ�ǡ�
��Һ��I��0.4����Ʒ��2mol��L������Һ��
��ƷO.1g�����Ҵ�l00ml�У���Ũ����lml��
����������ҺI�������������軯�ص�2mol��L������Һ��
���������ͪ��������ɫ������ȩ��Ӧ�����������ɫ���������ʻ������ﲻ��ɫ�� ���Ƶ���
��������ܲ���ȩ�ࡣ
��Һ��I��1%��5%�Ƶ����Ҵ���Һ��
��25%������炙�27%����������Һ��
������������ҺI��������Һ���
(4)�����
�������
�����л����ࡣ
��Һ�������0.1g�����Ҵ�500ml��0.1mol/L����������Һ5ml�� ���������塣
�������ɫ����������ɫ�ߵ㡣
�ڸ�����أ�����
����֬���������
��Һ����ͨ����ɫ�����Ը�����ء�
�۹�������
���������ᡣ
��Һ��0.3%����������Һ��
����������������(365nm)�¹۲졣
�������ǿ��ɫӫ�⡣
��2��6-���ȱ���-�����
�����л�����ͪ�ᡣ
��Һ��0.1%��Ʒ���Ҵ���Һ��
����������¡�
�������ɫ�����ʺ�ɫ��
(5)����
��Emerson �Լ�(4-����������֣����軯��(��))
�������ࡢ���㰷�༰�ӷ��͡�
��Һ��I��4-�����������1g�����Ҵ�100ml��
�����軯��(��)4g����ˮ50ml�����Ҵ�ϡ����100ml��
������������ҺI�����ȿ����и���5min��������Һ�������ȿ����и���5min��Ȼ���ú��а�����(25������Һ)���ܱ������С�
������ߵ�ʳ�-����ɫ���ӷ���������ɫ�����³ʺ�ɫ�ߵ㡣
��Boute ��Ӧ
�������ࡢ�ȡ��塢������ӡ�
����������������NO2����(��Ũ����)��������3��10min������NH2����(Ũ��Һ)������ ������(���ȴ��Ա���)
�������ࡣ
��Һ��1����Ʒ�ļױ���Һ��
��DDQ(���ȶ��������)�Լ�
�������ࡣ
��Һ��2����Ʒ�ļױ���Һ��
��TCNE (�������ϩ)�Լ�
�������ࡢ����̼�⻯��ӻ��ࡢ���㰷�ࡣ
��Һ��0.5%��1%��Ʒ�ļױ���Һ��
��Gibb��s(2��6-���屽�����ǰ�)�Լ�
�������ࡣ
��Һ��2%��Ʒ�ļ״���Һ��
���Ȼ���
�������ࡢ�������ᡣ
��Һ��1%��5%�Ȼ�����0.5mol��L������Һ��
������������ɫ����������ʺ�ɫ��
(6)����������
��FCNP(�����ƣ����軯��)�Լ�
����֬ ���庬������簱���衢�ҡ��������弰����������ἰ������ ��Һ��10������������Һ��10����������Һ��10�����軯����Һ��ˮ��1��1��1��3��ϣ����������ٷ���20min�����䱣�����ܣ���ǰ�����Һ���ͪ�������ϡ�
��Dragendorff(�⻯����Լ�)�Լ�
���������庬���������������ࡢ�����ɲ���ҩ�
��Һ��I����ʽ������0.85g����10ml�����ἰ40mlˮ�У�
�⻯��8g����ˮ20ml�С� ��
��������ҺI���������ϣ�����ɫƿ����Ϊ����Һ����ǰȡ����Һlml��������2ml��ˮl0ml��ϡ�
��������ٺ�ɫ�ߵ㡣
��4-��ɡ��ͪ
���������ӻ������
��Һ����Ʒ0.02g�����Ҵ�35ml����ˮ��100ml��
������������25����ˮ������������,ȡ�����������(365nm)�¹۲졣
�ܵⲬ���
����������༰�л��������
��Һ��10�����Ȳ�����Һ3ml��ˮ97ml��ϣ���6���⻯����Һ�����ȡ�����ǰ���ơ� ���������泥�����
����������������
��Һ��������1g������4mlˮ�У�����������1g����У���μ���Ũ����ֱ��������ʧ�� �����������1l0oC���������ӡ�
�����������ȡ���Ǯ�Ӽ��ˮ�ɼ��ڼ���ⶹ�����л��⻯����ܼ���� ��Ehrlich (�Զ��װ�������ȩ������)�Լ�
�������������P���ࡣ
��Һ��1����Ʒ��Ũ������Һ��״���1��1��ϡ�
������������50oC����20min��
������ʲ�ͬ��ɫ�İߵ㡣
(7)����
�����ᣯ�Ҵ�
����֬���尷�ࡣ
��Һ��50��65���������Ҵ�100ml�С�
��������Ҫʱ120oC���ȡ�
��2��6-���������ǰ�
����������������(������)��������֬�������١��巼�㰷������̼�⻯�ҩ�������������ݼ��ȡ�
��Һ�������Ʊ���0.5����2����Ʒ�Ҵ���Һ��
�����������110oC����10min��������������
������
�������ࡣ
��Һ��O.1����Ʒ���Ҵ���Һ��
�ܶ���ͪ��뿣��Ȼ���
�������ࡣ
��Һ��I������ͪ���1.2g������ˮ35ml�У����Ȼ���0��95g����ȴ���Ũ��ˮ2ml�� �������ǰ�0.12g����200mlˮ�С�
����������ҺI�����ϣ�����1�죬���ˡ�
��Pauly (����������)�Լ�
�������ࡢ�������ż�ϵ��ӻ������
��Һ������4��5g�������ȵ�12mol��L����45ml�У���ˮϡ����500ml��ȡlOml�ڱ�����ȴ����4��5��������������ҺlOml����OoC����15min����ǰ�ӵ����10��̼������Һ�� ����������(��)��
�������������١��尷�ࡣ
��Һ���������3g���Ȼ���1g����ˮ20ml��
�������ɫ���ۺ�ɫ�����ϳ���ɫ�ߵ㣬2h����ɫ���ˡ�����������ˮ����뱥��ˮ���������ڣ�������ɫ�㡣
��1��2-����-4-������
�������㰷�ࡣ
��Һ����Ʒ0.5g����95mlˮ��������5ml����ȥ�����T�á�
���������Ӧ30min��ɫ��
�������ǣ�����
�������㰷�ࡣ
��Һ��������2g����85������l0ml��ˮ40ml���Һ�У��ټ��Ҵ�����������30ml�� �����������115�����l0min��
(8)������������������
�٦�-����
����3��5һ��������������������������������
��Һ��I��O��5����-�����Ҵ���Һ��
��10���������ؼ״���Һ��
������������ҺI��������Һ��
������ʺ��ɫ�ߵ㡣
�ڶ��������Ȼ���
�����������ࡣ
��Һ��1.5���������Ҵ���Һ��0.1g�Ȼ��ٵ�0.2���Ȼ�����ҺlOOml����5��1��ϡ� ����: ����������(254nm)�¹۲졣
���������ɫ�ߵ㡣
(9)�����ἰ����
������ͪ
���������ᡢ���백�����ࡣ
��Һ����ƷO.2g�����Ҵ�l00ml�С�
�����������110oC���ȡ�
������ʺ���ɫ�ߵ㡣
������ͪ��������
���������ἰ�ӻ����ࡣ
��Һ������ͪ1g��������2.5g����l0ml�������У����Ҵ�ϡ����500ml�� �����������120oC����20min��
��1��2-����-4-������
���������ᡣ
��Һ������ǰ����ƷO.02g����5%̼����l00ml�С�
������������¸��
�������ͬ������ʲ�ͬɫ�㡣
�ܵ�죯����п
������������ijЩ���ࡣ
��Һ�����1g������п1g����95�������l00ml�У�������80oC����ȴ�������1ml�����䱣�档
�����������80��85"C����30min�� ��
������ͪ��������
�������ļ����ġ�
��Һ��1������ͪ�����Һ������ᰴ5��1��ϡ�
�����������l00oC����5min��
�����ȩ
���������ἰ���ࡣ
��Һ��I����Ʒ1g���ڱ���50ml��;
��1mol��L����������Һlml�����Ҵ�ϡ����lOOml��
������������ҺI����110oC����l0min��������Һ����110oC�ٸ���l0min���������(365nm)�¹۲졣
(10)����
�����ȩ������
�������弤�ء�
��Һ��1�����ȩŨ������Һ��
�����������105�����5min��
���Ȼ���
�����Ƽ����ࡣ
��Һ���Ȼ���0.2g���ں�����2ml�ļ״�60ml�С� ����������������(365nm)�¹۲졣
�۸�����
�������弤�ء�
��Һ��5��������״���Һ��
�����������110oC����5min���������(365nm)�¹۲졣 �����Ȼ��࣯����
��������������ࡣ
��Һ�����Ȼ���20g��������20ml���ȷ�60ml���Һ�С� �����������100oC����5min������ⳤ���¹۲졣 �����������ߵ�ʺ��-����ɫ��
�ݶԼױ�����
�������廯�����ͪ����������ࡣ
��Һ��20����Ʒ���ȷ���Һ��
�����������100oC���������ӣ�����ⳤ���¹۲졣 ������ߵ��ӫ�⡣
���Ȼ��ᣯ����
�������ơ��������廯���
��Һ���Ȼ���5ml����ȴ�¼�����l0ml�ܽ⡣
�����������130oC����5��l0min��������ⳤ���¹۲졣
������ߵ���ӫ�⡣
(11)����
�����㰷���ڱ������Լ�
����̼�⻯���
��Һ��1.23g���㰷��1.66g�ڱ�������l00ml 95���Ҵ��е���Һ��
��������������ա�
��������dz���ɫ�������dzʻ���ɫ�����dz���ɫ����ȩ�����ɫ��
��������Ǧ��2��7һ����ӫ����
����߰�ࡢ���ࡢ������
��Һ��I��2��������Ǧ�ı�������Һ��
��1��2��7һ����ӫ�����Ҵ���Һ��
ȡ��ҺI���� ��5ml���ȣ��ø���ı���ױ�ϡ����200ml���Լ���Һֻ���ȶ�2h�� ���������塣
���ڰ�������������
�������ࡣ
��Һ��O.3g�ڰ���������85������5ml���Ҵ�95ml��
����������llOoC����15��20min��
������ߵ�ʺ�ɫ��
�ܱ�����������������
������ԭ�ǡ�
��Һ��4g��������4ml������20ml 85�����Ṳ����200ml��ͪ�С�
�����������85�����l0min��
���������������ɫ��1��4-��ȩ�ǡ��;��dz���ɫ��
��˫��ͪ������
����ͪ�ǡ�
��Һ��˫��ͪ(5��5-����������-1��3-��ͪ)10.3g����90ml�Ҵ���l0ml 85�������С� ������������110oC����15��20min��
������չ��¹۲죬��ɫ�����ϳʻ�ɫ�ߵ㣬����ⳤ���³���ɫӫ�⡣
������������������
�������ࡣ
��Һ��0.5g����������l0ml���ᣬ�ټ�10ml40����������ˮ��Һ�����Ҵ�ϡ����l00ml�� ����������������������15min��
������ߵ�ʻ���-���ɫ��
�߶Զ��װ�������ȩ��������ͪ
�����������ࡣ
��Һ��I. 5ml 50������������Һ��20ml�Ҵ����ȣ�ȡ����Һ0.5ml����������ͪ0.5ml��������50ml�Ļ��Һl0ml����������Һ�����������ƣ�����ǰ��ϣ�
��. 1g�Զ��װ�������ȩ����30ml�Ҵ��У��ټ�30mlŨ���ᣬ��Ҫʱ����Һ����������180mlϡ�͡�
����������I����105oC����5min�������Ȼ����90�����5min��
������ߵ�ʺ�ɫ��
(12)ɱ���
�ټ���
�����Ȼ�ɱ��������������
��Һ��O.1g������70ml�Ҵ��У���25mlˮ�����Ҵ�ϡ����l00ml��
���������������¸���������˹�Ƭ�����������5min��
�������ɫ�����ϳʺ�ɫ�ߵ㡣
���壯���Ȼ�̼
�����л���ɱ�����
��Һ��10��������Ȼ�̼��Һ��
��������������
���̣�ˮ��ȩ
�����л������ɱ�����
��Һ��I��100mg�Ȼ�������100ml 80���Ҵ���
���ܽ�1.3g 2-�������С�����Ҵ��У���1gˮ��ȩ��5ml�Ҵ�����1��2�α����ᣬ�ϲ�����Һ������30min����ȴ������ˮ��-2-ȩ-2-���������������Ҵ��ؽᾧ����50mg������������100ml�Ҵ��С�
������������ҺI�����Ϻ���塣
�ܶ��������Ȼ�п
�����Ȼ�ɱ���(DDT��CPCA����-DDT���˾����������ȡ���ɱ��)��
��Һ�����������Ȼ�п��0.5g����100ml��ͪ�С�
���������200oC����5min��
���壯ӫ���أ�������
����ɱ�����
��Һ��I��5��������Ȼ�̼��Һ��
��1ml 0.25��ӫ���صĶ�����������Һ�����Ҵ�ϡ����50ml��
��1.7g����������5mlˮ�У���2-�������Ҵ����ñ�ͪϡ����200ml��
������չ����ı�������ҺI������30s��ȡ��������Һ��������Һ�������������7min��
(13)��ͪ��
���Ҵ�������������
������ͪ�ࡣ
��Һ��I. 1���Ҵ������������εļ״���Һ��
��. 5�����Ҷ������Ҵ���Һ��
������������ҺI��������Һ��ʹӫ���ȶ�����������ⳤ��������2min��
�����������¹۲�ӫ�⡣
�����Ȼ���
������ͪ�ࡣ
��Һ��10�����Ȼ�����ȷ���Һ��
��������塣
���������ⳤ���³�ӫ�⡣
�����Ȼ���
������ͪ�ࡣ
��Һ��1�����Ȼ����Ҵ���Һ��
��������塣
���������ⳤ�����Ի�ɫӫ�⡣
��Benedict(����ͭ����������)�Լ�
�������ڶ��ǻ��Ļ�ͪ�༰�㶹���ࡣ
��Һ��1.73g ����ͭ(CuS04?5H20)��17.3g �������Ƽ�10g ��ˮ̼��������ˮ��ϡ����1 00ml��
��������塣
���������ⳤ���¹۲죬���ڶ��ǻ��Ļ�����ߵ�ӫ����������
(14)������
��������
�����ϻ�-SH��(���װ���)��˫���-s-s-��(�װ���)�������ᡣ
��Һ��I. 1.5g ����������5ml 2mol��L������Һ��95ml�״��У���10ml 25�����������Һ�����ˣ����ã�
��. 2g�軯������5mlˮ���ü״�ϡ����100ml��
������������ҺI��������Һ��
������ϻ�������ʺ�ɫ���������ɳȱ�Ϊ����ɫ��˫�����ڻ�ɫ�������Ժ�ɫ�� ��FCNP(�����ƣ����軯��)�Լ�
����֬���庬��̨��簱���衢���弰�������
��Һ��10������������Һ��10����������Һ��10�����軯����Һ��ˮ��1��1��1��3��ϣ���ǰ��������ͪ��ϣ���ʱ�������ơ�
��3�Ȼ������ǻ�ˮ����
��������������ࡣ
��Һ��I��1���Ȼ�����80���Ҵ���Һ��
��1���ǻ�ˮ�����80���Ҵ���Һ��
����������������������10min������ҺI������15min��������Һ��
�������ɫ�����ϳʰ�ɫ�� �� ��
�ܵ�������
���������ᡢ��������ù�ء�
��Һ����������Һ-������3g �ܺ�100ml 0.05mol��L����Һ��(�������ƣ�����������б�ը��)��
�ݵ�죯����
������������
��Һ��0.4g�������100mlŨ�����С�
����������120oC���ȡ�
������ʲ�ͬ��ɫ��
(15)��֬������
��������
�������ᡢ��֬���֬���ἰ���ࡣ
��Һ��250mg ��������50ml�Ҵ��С�
������������120oC���ȡ�
������6G
������֬���
��Һ��lg ����6G����100ml��ͪ�У�
���������������ⳤ���¹۲졣
����������
������֬���
��Һ��O��04g������������100ml O��01mol��L����������Һ�С�
(16)������
��˫����
�����������ӡ�
��Һ��20mg˫��������100ml��ͪ�У�����ɫƿ���ڱ����ڱ��档 ������������Һ��������25�����������Һ��
�ں찱�� (�����������)
�����ؽ������ӡ�
��Һ��0.5���찱���Ҵ���Һ��
����������������Һ���Ըɺ��ñ����ڰ������С���
������
�����ڶ������ӡ�ϡ�����ˡ�
��Һ�������Ҵ�������Һ��
����������ֱ�ӷ��백�����С�
�������軯��
����Fe3+��Cu2+��Cl��Mo��V��W���ӡ�
��Һ�������Ƶ�2�������軯����Һ��
�ݶ���������
����Ag+��pb2+��Hg2+��Cu2+��Sn2+��Mn2+��Zn2+��Ni2+��Co2+��Ca2+���ӡ� ��Һ��I��1����2�������������Ҵ���Һ��
��25�����������Һ��
������������ҺI��������Һ��
�����Ni2+����ɫ��Co2+�ʳȺ�ɫ��Zn2+�ʺ���ɫ��Ag+��pb2+��Cu2+��Sn2+��Mn2+�ʺ�ɫ�����ṯ�ӳ�����80oC����Ƭ�̣��ߵ����ɫ��
(17)������
��������
�������������ӡ������Ρ��������������Ρ��������μ��������±�ء�
��Һ��0.1��0.5mol��L��������Һ�мӰ�ˮֱ�������ܽ⡣����ǰ���ơ�������γ��ױ����ʡ�
�����������110��120oC����l0min����Ҫʱ���������l0min��
����������������泥�ӫ����
����±�����ӡ�
��Һ��I��1g����������l00ml O.5mol��L���������Һ�У�
��1gӫ��������l00ml�Ҵ��С�
������������ҺI���Ըɣ�������Һ��
������泥���������
������������������������ ��
��Һ��I��5������淋�lmol��L������Һ��
��5������������Һ��
������������ҺI��������Һ����105�����30min��
��������P��������γ�����ɫ�������γ�����ɫ��
�ܷ�ľ���
����������������������
��Һ��I��0.02����ľ����lmol��L������Һ��
��2mol��L����������Һ��
������������ҺI��������Һ��
����������Ρ������γ�Ѫ��ɫ�������γʳȻ�ɫ��
���ģ�.����������ר����ɫ����
�⣺
�����ڲ����ͻ��߷����廯����
���Ʒ�������100ml ���ƿ�У�����һ����ֽ������������
������ƿ�У�����10g ������30g �轺
�������
�����ں���ԭ�Ի��Ż���������ǻ���������ȩ
���Ʒ�����1.5g KMnO4 + 10g K2CO3 + 1.25mL 10% NaOH +200mL ˮ. ʹ����3 ����
������(PMA)
����
���Ʒ�����10 g of ������+100 mL �Ҵ�
�����
�����ں�������ŵĻ�������㻯����
�����棺
�����
���Ʒ�����10%������(IV)+15%�����ˮ��Һ�Ȼ���
���������
���Ʒ�����1% FeCl3 + 50% �Ҵ�ˮ��Һ.
ɣɫ��(�ǻ���ͪ)
����, ��ӫ�����
���Ʒ�����0.1% ɣɫ��+�״�
����ͪ
�����ڰ�����
���Ʒ�����1.5g ����ͪ+ 100mL of ������+ 3.0mL ����
����������(DNP)
������ȩ��ͪ
���Ʒ�����12g ����������+ 60mL Ũ����+ 80mL ˮ+ 200mL�Ҵ� ���ȩ(������)
����
���Ʒ�����15g ���ȩ+ 250mL �Ҵ�+2.5mL Ũ����
�����
���������ᣬpKa<=5.0
���Ʒ�������100ml �Ҵ��У�����0.04g ����̣������μ�0.1M��NaOH ˮ��Һ���պó�����ɫ������
������
����
���Ʒ�����235 mL ˮ+ 12 g ���ᰱ+ 0.5 g �����氱+ 15 mLŨ����
����ȩ(�Լ���������ȩ)1
����
���Ʒ�����135 �Ҵ�+ 5 mL Ũ����+ 1.5 mL of ������+ 3.7 mL����ȩ�����ҽ��裬ʹ��Ͼ���.
����ȩ(�Լ���������ȩ)2
��������ϩ��������(cineoles), withanolides, ���̼�(acronycine)
���Ʒ���������ȩ:HClO4:��ͪ:ˮ(1:10:20:80)������Ҫ�����ܼ��ļ��Դ�С����չ�����ļ������Ե�����
�ġ�����������������ע������
��һ���̰�
�̰��õ��Ƚ����˹������ϡ�������������׳����϶���ͣ����ɵIJ��ƣ���ϡ��ˮ���������ϴֲڡ��Ƚ����һ���ǹ轺G:ˮ=1��2��3���轺G:�ȼ���ά����ˮ��Һ=1:2����ĥ�Ƚ���ʱ�䣬���ݾ��������������ʪ���йأ�һ��ͨ�������а�ʱ�Ƚ��µε�������жϣ�Խ��Խ���µΡ��Ƚ���ϡ����Ӱ����ƽ���⣬ҲӰ���Ϳ��ĺ�ȣ���һ��Ӱ����������Ϳ�㱡���������أ�Ϳ�����ɫ����ô���ԡ�ͨ������������Ա�������Ӱ�첻�Ǻܴ�Ӱ��������չ���������ƺ�չ��ϵͳ�ı��͡�
��������
������С�ĵ����ܡ�������㹻�����ԣ����ֻ��1���ĵ����ܡ���������İߵ��С��չ����ɫ��ͼ����Ⱥã���ɫ��������Ʒ��Һ�ĺ�ˮ��ԽСԽ�ã���Ʒ��Һ��ˮ�������ߵ���ɢ����Ʒ��Һ���ܼ�һ������ˮ�Ҵ����״����ȷ¡�����������������ı�����õ紵����ȷ紵�ɻ��������������ɡ�
������չ��������
ѡ����ʵ������Ѹ�����ܼ������Һ©����ǿ����ҡʹ���Һ��ֻ��ȣ����ã�����ֲ㣬
ȡ��������һ����Ϊչ���������Բ�Ӧ�ðѸ������Һ����չ���ף���ҡչ����������չ��������ϲ����Ⱥ�û�з�Һ��չ����������ɲ�������ȫʧ�ܡ�������ܼ��ı���ȷ�ȶԲ�ͬ�ķ��������в�ͬ��Ҫ�����ﵽʵ������������߾�ȷ�ȣ����磺ȡ1ml���ܼ���Ӧʹ��1ml�ĵ�����Һ�ܣ���Һ��Ӧ���ϼ�����֤Ҫ���ܶ���ʱ���ⲻ�DZ���ġ�
���ģ�չ��ϵͳ�ı���
һ��ʹ�õ���˫�۵�չ���ף�һ��������չ��������һ�ۿɼ��백ˮ�����ᡣ�Ѵ�չ���İ�������ۼ��ƽ̨��б���ţ�����չ���ĸ��ӡ���չ��������������չ���ף���ʹ��������������ﵽ���ͣ���ֹ����ЧӦ������ʱ���ڰ��Сʱ���ҡ�չ��ʱ����Ҫ���Ӱѱ�������չ�����У������Ա����������������ƽ��Ӱ�첻��Ȼ����Ӧ�þ����ᡢ�졣 ��ʪ�ȵĿ�����ʪ�ȶԱ���Ӱ�춼�ܴ������ǰ���£�ͨ���¶�Խ�ͷ���Խ�ã����ѵķ������ڵ����·��룬�����˲����ա�ʪ�ȵ�Ӱ�죬������Ҫ��Ӱ�챡������������������ѡ���ԣ��������ӣ��ı仯��ʪ��Ӧ����ʵ�����ȷ�����¶ȿ���ʹ�ÿյ��������ʪ�ȿ�����ͨ������һչ���۷�����ӦŨ�ȵ����ᡣ
���壩��ɫ��
����ɫ����ɫ����Ҫ���кõ�������
�塢TLC�мǵ�Ҫ��
��һ����ij����Ʒ������չ������ֻ��ʾһ���㣬���������ڱ��չ������Ҳֻ��ʾһ���㡣�����Ѱ��չ����ʱ���ೢ�Լ��ֱ�����ͬ���ɷֲ�ͬ��չ������չ�����ļ���̫С����ֲ���������̫��Ҳ�ֲ���.һ����Ŀ������Rfֵ��0.3����Ϊ��ѡ�
���������㲻�ܵ��̫Ũ,���������ص�,�����ж�,��Ϊ�������������Ļ�,һŨ�ͱ��һ�����ˡ�
�����������ϵ��չ���������̶Ⱥ��ܼ��ļ��Ժ������ڸ��ܼ��е��ܽ����йأ�ֻ�����߱ȽϺ����ˣ�������һ�����õķ���Ч����
ѡ���ʵ���չ��������Ҫ����.һ�㳣���ܼ����ռ��Դ�С�����˳�����д��Ϊ��ʯ����<����<��<����<THF<��������<��ͪ<�Ҵ�<�״�&NBSP; ʹ�õ�һ�ܼ����������ܴﵽ
�ܺõķ���Ч��������ʹ�û���ܼ�ͨ��ʹ��һ�����Ժ͵ͼ����ܼ���ɵĻ���ܼ������Ե��ܼ������������ֶȵ����ã�
���õ��ܼ�����У�
Petroleum ether/Ethyl acetate, Petroleum ether/Acetone, Petroleum ether/Ether, Petroleum ether/CH2Cl2, CH2Cl2/ethyl acetate, ethyl acetate/ MeOH, CHCl3/ethyl acetate
չ�����ı���Ҫ������.һ����������б����ĸ��������ʲô����չ�����������ȳ���ʹ�ø���չ������Ȼ�ϳ��Ա�����ֱ���ҵ�һ������Ч���õ�չ�������ܶ�ʱ��,չ������ѡ��Ҫ���Լ����ϱ任չ������������ﵽ���Ч��������ʵ���У�Ϊ��ʵ��һ������������������Ч���룬�������������е��ܼ�����������Ϻ������ҵ�petroleum ether/THF��������Ļ���ܼ���
һ��������ܼ����ʱ,���ø���/�ͼ��Ե������Ϊ1/3�Ļ���ܼ�,����зֿ��ļ���,�ٵ�������,�ﵽ���Ч�������û�зֿ��ļ�������ǻ��ܼ���
�����ڹ轺�����������������ֽ�����ʣ���չ������������һ������Ұ�����ˮ����वȼ����������к轺�����ԡ���Ȼ��ѡ�������ӵļ������ʣ������뿼�����״Ӳ�Ʒ�г�ȥ����ˮ�����ǽϺõ�ѡ��
����Ҫ�ر�ǿ����һ���ǣ�����Ч���ĺû������ù轺���ܼ����������й�ϵ�� ��ͬ���������Ĺ轺���ܺ�ˮ���Լ������Ĵ�ϸ�̶ȣ�����ǿ����ͬ��
�Ӷ����²�Ʒ��ij�����ҵĹ轺�з���Ч���ܺã�������һ�����ҵľͲ��С�
�ܼ��ĺ�ˮ�������ʺ����Է���Ч���������Ե�Ӱ�졣�¶ȶԷ���Ч��Ӱ��Ҳ�����ԡ�����ʵ����û�пյ�������ʵ����̷��֣������������ͬ�IJ�Ʒ���������ϴ������Ҫ���ڶ���С�ܶ࣬��һ����Ҳ�ǿ�������ġ� ���ڴˣ�ʹ�õĹ轺������ʱһ��Ҫ�ܷ⣬��ֹ������TLC���õĹ轺��һ��Ҫ�����ڸ��������棬��ʹ��ǰ�ں�����������һ��ʱ�䡣 ������TLC���ٷ�Ӧʱ���ڵ���ʱ�������Ƿ�Ӧ��ϵ�Ļ����Һ��һ����A��ÿ��
�ѻӷ���ԭ�ϸ���һ����B,C, D�ȵȣ�Ȼ�����е�ԭ�Ϻͷ�Ӧ��ϵ�Ļ���ܼ��ٹ�ͬ��һ����ϵ�X����Щ���ڰ��ϵ�λ����ͼ��ʾ��
�����ĺô���չ�����������ؿ���ÿ�����λ�ã���A�����չ����ĸ�����ݵĵ���B,C,��ԭ�ϱȽϣ��Ӷ��ж�ԭ����ʧû�У����ϵ�X��Ŀ�����ڣ�����۲죬��Ϊ��ʱ��չ�������λ����Щ���Σ��������ڱ�ԵЧӦ�ȵȣ�ʹ���жϲ��ס�
����TLC�ж����ʵĴ���ʱ������Ҫ��NMR���ϣ���Ϊij����Ʒ������չ������ֻ��ʾһ���㣬���������ڱ��չ������Ҳֻ��ʾһ���㡣����Ȥ���ǣ�����H1NMR�ɼ���Ĵ���Ҳ����95%���ң���ʱ��H1NMR��ʾ�ϴ��Ķ�����һ���ͻᷢ���м����㡣���ԣ�����Ҫ���ʹ�á������Լ���ǰ����Ʒ�������ֻͨ��������жϴ��ȡ�
���ģ��������¼�������
1�� �����ڲ������ͱ���IJ�ͬ����ʹ����ʹ�õĹ轺����ͬ�������ڰ�TLC�����õ���չ��������������ʱ��Ҳ�Եü���ƫ������Ҫϡ��һ���������ʹ���ֹ��̵�TLC���������ؽ�ϴ�Ѽ���ϴ��ǿ�ȼ�Сһ��������˵����������ʹ��Ԥ�Ƹ�Ч�壨HPTLC���������
2�� �������ڹ轺�����������������ֽ�����ʣ���չ������������һ������Ұ�����ˮ����वȼ����������к轺�����ԡ���Ȼ��ѡ�������ӵļ������ʣ������뿼�����״Ӳ�Ʒ�г�ȥ����ˮ�����ǽϺõ�ѡ����Ӧ���������㻹��TLC�������������Ӷ��Ұ��ȽϺã����ڷ���TLC�������Ұ����ð�ˮ�����Ǽӵ�չ����������ԡ�
չ������ѡ��
���л��ϳ�ʱ�߰����dz��е��£�չ������ѡ���������Ҫ�ˡ�
ѡ���ʵ���չ��������Ҫ����.һ�㳣���ܼ����ռ��Դ�С�����˳�����д��Ϊ��ʯ����<
����<��<����<THF<��������<��ͪ<�Ҵ�<�״�ʹ�õ�һ�ܼ����������ܴﵽ�ܺõķ���Ч��������ʹ�û���ܼ�ͨ��ʹ��һ�����Ժ͵ͼ����ܼ���ɵĻ���ܼ������Ե��ܼ������������ֶȵ����ã�չ�����ı���Ҫ������.һ����������б����ĸ��������ʲô����չ�����������ȳ���ʹ�ø���չ������Ȼ�ϳ��Ա�����ֱ���ҵ�һ������Ч���õ�չ������չ������ѡ���������ٶԵ�����ɷ������õ��ܽ��ԣ��ڿ�ʹ�ɷּ�ֿ����۴�����ֵ�Rf��0.2~0.8֮�䣬�����ⶨ��0.3��0.5֮�䣻�ܲ��������ֻ�������������ѧ��Ӧ���ݷе����У��Ƚ�С����չ������ְߵ�Բ�Ҽ��У�����ܼ�������������ơ� һ����˵���������ܼ���ϵ�Ļ����������������ˮ��ɣ��ٸ�����Ҫ����״����Ҵ������������������ܼ�ϵͳ�ļ��ԣ��Դﵽ�õķ���Ч�����ʺ���������ͪ������ȵķ��룻�еȼ��Ե��ܼ���ϵ���ȷº�ˮ����������ɣ��ɼ״����Ҵ������������������ڣ��ʺ����������㶹�أ��Լ�һЩ���Խϴ��ľ֬�غ�����ķ��룻ǿ�����ܼ�������������ˮ��ɣ�Ҳ���״����Ҵ������������������ڣ��ʺ��ڼ��Ժܴ������������ķ��롣�ܶ�ʱ��,չ������ѡ��Ҫ���Լ����ϱ任չ������������ﵽ���Ч����������ʵ���У�Ϊ��ʵ��һ������������������Ч���룬���������˺ܶ��ֵ��ܼ���ϣ������ҵ�ʯ����
��EtOAc��HCOOH��5.5��3.5��0.1������ܼ���һ��������ܼ����ʱ,���ø���/�ͼ��Ե������Ϊ1/3�Ļ���ܼ�,����зֿ��ļ���,�ٵ���������������������ܼ���,�ﵽ���Ч�������û�зֿ��ļ��ߵ�ϡ��ϡ���������ǻ��ܼ��������ڹ轺�����������������ֽ�����ʣ���չ������������һ������Ұ�����ˮ����वȼ����������к轺�����ԡ���ѡ�������ӵļ������ʣ������뿼�����״Ӳ�Ʒ�г�ȥ����ˮ�����ǽϺõ�ѡ������Ч���ĺû������ù轺���ܼ����������й�ϵ����ͬ���������Ĺ轺���ܺ�ˮ���Լ������Ĵ�ϸ�̶ȣ�����ǿ����ͬ���Ӷ����²�Ʒ��ij�����ҵĹ轺�з���Ч���ܺã�������һ�����ҵľͲ��С��ܼ��ĺ�ˮ�������ʺ����Է���Ч���������Ե�Ӱ�졣�¶ȣ�ʪ�ȶԷ���Ч��Ӱ��Ҳ�����ԣ���ʵ�������Ƿ�����ʱͬһչ���������������Rf��Ȼ��ͬ ��
�@ չ������ѡ����Ҫ������Ʒ�ļ��ԡ��ܽ�Ⱥ��������Ļ��Ե����������ǣ��ڽ��б������ʱ������Ӧ��֪��δ֪��ѧ�ɷֵ����ͣ��伫�ԵĴ��¹���������ȡҺ���ɫ�����������༫�Կ�֪������ij��Ʒ�ﺬ���ֻ�ѧ�ɷ��Ȱ����Բ�ͬ���·֣�Ȼ��ϸ�֣����ڷ���δ֪�Ļ�ѧ���ʣ�չ������ѡ��Ҳ��һ�������Ĺ��̣���Ӧ�ý�����չ�������ǣ��������ۺ�
�������ܼ��������������ܼ���ѡ����ַ����ϵ������������ʱ���õ��ܼ�����һ�������ܼ���ϰ���ϳ�ϴ�Ѽ������ڱ����ֽ����ʱ����չ������ϴ�Ѽ���ѡ������ݱ�������������ѡ�õ����������������߽���������Կ������ü������������в���ʱ��������������Ϊ���������ʣ�һ��ѡ���������ܼ�Ϊϴ�Ѽ�������������Ϊǿ���Գɷ֣�����ѡ�ü����ܼ�Ϊϴ�Ѽ��������ijһ���������������Խ����������������Թ�������ʯ�۴���轺������ϴ�Ѽ��ļ���������Ӧ���͡����������ʱ����������Ʒ�ڼ���ʱ�ɲ����ڷ������ѡһ���˵��ܼ�����Ʒ�ܽ����롣�ܽ���Ʒ���ܼ�Ӧѡ���Խ�С�ģ��Ա㱻����ijɷֿ��Ա�������Ȼ�������ܼ��ļ��ԡ����ּ��Ե�������һ��ʮ�ֻ����Ĺ��̣���Ϊ���ݶ�ϴ�ѡ���ʹ�����ڲ������ϵĸ����ɷ������ϴ�ѡ������������������ݶ�̫���Ͳ��ܻ������ķ��롣�ܼ���ϴ����������ʱ�������ܼ��Ľ�糣�����ţ�����ʾ����糣���ߣ�ϴ�������ʹ����ϵ�ϴ��˳��������ڼ�������������轺�����������ԷǼ����������������̿��������������˳���෴����ˮ����ˮס�ܼ������γɵ��������ã�����֬�����ܼ���Ϊǿ��
���������ʵ�����
���������������������ϴ�Ѽ���ͬ�������������е�����Ҫ�أ��˴˽�����������ָ������������ϴ�Ѽ��������£������ɷֵķ��������ֱ���뱻�������ʵĽṹ�������йء��Լ������������ԣ��ɷֵļ��Դ�����סǿ����Ȼ���в�ҩ�ɷֵ�������ӹ�����Ҫ�ģ����缫�Ի��ŵ���Ŀ���࣬��������ס�ܾͻ����Щ����ͬϵ����̼ԭ����Ŀ��Щ��������Ҳ��ǿЩ����֮��ֻҪ�����ɷ��ڽṹ�ϴ��ڲ�𣬾��п��ܷ��룬�ؼ�����������ѡ��Ҫ���ݱ��������ʵ����ʣ�������������ǿ�ȣ����ܼ������������ߵ����ϵ�����ǡ�����Ҫ���DZ��������ʵļ��ԡ��类�������ʼ��Ժ�СΪ����������ϩ�����京�����Ǽ��Ի��ţ�����ѡ�������Խ�ǿ���������������������ܼ���ʯ���ѻ���ϴ�ѡ���������ҩ�ɷֵļ��Խϴ�����Ҫѡ���������ܽ�������������һ������������õ�ϴ�Ѽ�����Ӧ��С����ijһ�ݶȵ��������Ӧ�ñ���������жϱ���������ij���ܼ�ϵͳ�еķ�����������⣬�ܷ�������ķ��룬����ѡ����ܼ��ݶ��кܴ��ϵ������ʵ��˵��������������������ϴ�Ѽ�����Ʒ����֮��Ĺ�ϵ�����ж���ֵĻ�����ֲ����֬ϵ��������ϩ����Ҩ�����ࡢ���������֬�������ݡ������C-27������߰Ԫ��ɷ֣�������������ǻ�
��Ŀ�Ķ��ٶ���÷��룺�������߰Ԫ���ں���5���ȷµı��У��������������������ϣ��������µ��ܼ������ݶ�ϴ�ѡ�����������Խ����Ĺ���þ����������������ڹ���þ�������Խ�����ϴ�Ѽ��ļ�������Ӧ���ͣ��༴���ñ���5���ȷµı������ɽ�һԪ�ǻ���߰Ԫ����������ϴ����������һ����˵����ͬ�����в�ҩ�ɷ��ڲ�ͬ���������в���ʱ�����ò�ͬ���ܼ����ܴﵽ��ͬ�ķ���Ч�����Ӷ�˵�����������ܼ���������ɷ����ߵ����ϵ��
������������������������һ�ּ�㡢���١����IJ���������һ�㽫�������õ�������������ƽ���粣��Ƭ�ϣ��γ�һ������в���ʱһ���Ʊ����������ԭ�����������������ơ�
1������������ص㣺���������Ӧ�������������ص����������ıȽϡ�
2����������ѡ��������õ�����������ѡ��ԭ�����������ͬ����Ҫ�������ڱ������Ҫ����������֧�ּ��������ȸ�ϸ��һ��ӦС��250Ŀ����Ҫ�����Ⱦ��ȡ����ڱ����������������Ԥ�Ʊ���һ���Ȳ��˹��ߣ��Ԣ�Ϊ�ˡ���չ���������污������ȴ�ϸ��������������Խϸ��չ��������Ӧ���̣�һ�㲻����10���ף����������ɫ����ɢӰ�����Ч����
3��չ������ѡ��������������������Ϊһ��ֵʱ����������Զ���ֵ���Ʒ�ܷ�������ķ��룬������չ������ѡ���в�ҩ��ѧ�ɷ���֬���Գɷ��У����¿ɰ��伫�Բ�ͬ����Ϊ���ԡ������ԡ��м�����ǿ���ԡ�����ʵ�ʹ����У�������Ҫ�����ܼ��ļ��Դ�С����չ�����ļ������Ե�����
����ɫ��С֪ʶ
1��������ߵ���Ч�ʣ� ���������TLC����������Ҫ��Դ��
ʵ��֤��������ëϸ�ܸ��ʺϽ�С����ĵ�������ע�������ʺϽϴ�����ĵ���������Ҫ����Ϊ��ע������С���ݡ���Һ���������Ӱ��ϴ�Ϊ���ⲻͬ����ëϸ�������Ƶ�ռ��������һ�鱡����������ͬһֻ����ëϸ�ܡ���Ӧע�������Ʒʱ��Ӧ��ëϸ���ó�������ͬ�����ܿμ���ϴ�ɾ������Ʊ���Ʒʱ�������ܼ��Ȳ��ܹ��ߣ��Ա��ڵ������ܼ��е�������������ױ䣬�������ı�չ������ɣ�����Ʒ�ܽ�ȹ���ʹ���㷢�����������õ��ܼ�Ϊ�״����Ҵ�����ͪ������TLC����ԭ��һ��Ϊֱ��3mm������
1-2cm�ױ߾���1.5cm;HPLC����ԭ��һ��Ϊֱ��1mm����5mm�ױ߾�1cm.
2��չ������ѡ��
TLC��HPLC��ȣ�һ��ͻ�����ŵ�����������ѡ����и��������ԣ��������ѡ���Ŀ����ʹ������Ʒ��Rfֵλ��0.1��0.7֮�䲢�ﵽ�Ϻõķ��룬��˶�Ӧ����������Ҫ���ʵ���ǿ�Ⱥ���ɡ�������ǿ��Խ������RfֵԽ���ܿ��ܽ��ͷ������������ⵥһ�ܼ����ѷ���ϸ��ӵĻ���������������ԭ����Ҫʹ�ö�Ԫ�ܼ���ϵ��һ��չ������ϵѡ�����£����ݷ�����Ʒ���ʡ�����ɫ�װ�����ѡ��һ����Ԫ�ܼ���ϵ��ͨ�������ܼ�������Ѱ���ʺ�Rfֵ���ʺϵ�Rfֵ�ҵ�����Ѱ���Բ�����ͬ�Ķ�Ԫ�ܼ���ϵ�����������������Ϊ�������һ�������Σ���ɿ����������ζ����Ƕ�Ԫ��ϵ��������Ԫ��ϵ��������������Ԫ��ϵ�����Ҽ���һ�£��ɸ��ݼ���ԭ���ó���һ����ɣ����ַ�����Ϊֱ�ۣ�Ҳ�ϼ�
3��TLC��ͨ����ɫ����
�������ɫϣ�������ȸߣ��ߵ���ɫ�ȶ����ߵ��뱳����ĶԱȶȺã��ߵ�Ĵ�С����ɫ����������ʵ��������ȣ�����Ʒ��ɲ�����ȫ��֪������£�ͨ����ɫ�����Ե���Ϊ��Ҫ��ͨ����ɫ������Ҫ�У�
1���������䷨�����㡢���ƻ���Ʒ��
2������������ͨ����ǿ�������ⷨ��������ȸ��ڸ���������ʹ�ã�
3��ӫ���Լ�������ӫ�ⱳ����ʹԭ����������ӫ�����ʱ�������ӫ�����ʸ����ԣ�
4�������ܼ����Ծ�������л�����Ч�������ƻ��ԡ�
- ��ɫ����ʵ�鱨��
-
��ɫ�ͱ���ɫ��----Ԥϰ����
Ԥϰ����һʵ�����Ʊ���ɫ����ɫ��ʵ��Ŀ��1ѧϰ����ɫ����ɫ������ԭ����Ӧ��2���ձ���ɫ����ɫ���뼼���Ͳ���Ҫ��3�����硭
- ��ɫ���뱡��ɫ��
-
ʵ��ʮ ����ɫ����ɫ��
ʵ��ʮ����ɫ����ɫ��12ʵ��Ŀ���˽ⱡ��ɫ����ɫ�Ļ���ԭ��ѧϰ�ñ���ɫ����ɫ���봿���л�������ļ���ʵ��ԭ��ɫ���Ƿ��롭
-
����ɫ��ʵ�鱨��
�л���ѧ�ڶ�����ʵ�鱨��һ������Ϣ�����꼶20xx��רҵ��ζӱ�����20xx��5��23��ʵ�����л���ѧʵ���Ҷ���ʵ�鱨������ʵ���⡭
-
��ʿ��ȹ����ܽ�
���ؽ�ȡ���ڲ����ܽ����Ȳ���������XXX���չ����ܽ�20xx�꣬���ۿ��쵼�Ĺ��ġ����ӣ��ڻ�ʿ����֧���£���ȫ�ƻ�ʿ�İ����£�Բ����
-
20xx���ز����۹����ܽ�
20xx�깤���ܽ�20xx�����Ͼ�Ҫ��ȥ�ˣ���ȥһ����Լ���˵����һ���ɳ����ջ��һ�꣬ת�ۼ�����˾�Ѿ�һ����ˣ��ս��뷿�ز���ҵ��
-
20xx���ϰ�����������ܽ�
����ú��20xx�����רҵ�ϰ��깤���ܽἰ�°��갲��һ��20xx���ϰ������������չ��������ϰ����ڼ̳�ȥ�깤������ɾ��Ļ����ϡ�
-
��ѧ��ѧУ��ȫ�����ܽ�
20xx-20xxѧ�����ѧ�ڣ���У���ϼ��йز��ŵ�ֱ���쵼�£����Ԥ��Ϊ�������ν�ϡ���ǿ������Ⱥ��Ⱥ�ε�ԭ��ͨ����ȫ����������
- ����˼��Ʒ�½�ʦ���˹����ܽ�20xx.1