阿司匹林湿法制粒压片及含量测定实验报告
阿司匹林颗粒剂的制备及含量测定
学生姓名
专 业
班 级
指导教师
阿司匹林 (Aspirin),又名乙酰水杨酸、醋柳酸 (Acetylsalicylic Acid),其化学名为2一(乙酰氧基 )苯甲酸,化学式为C9H8O4,是水杨酸的衍生物,白色结晶或结晶性粉末,无臭或微带醋酸臭, 味微酸。微溶于水 (1:3000),其饱和水溶 液加FeC1 试液即生成紫红色 ,阿司匹林易溶于乙醇(1:7),可溶于乙醚(1:20),氯仿 (1:17)。熔点为135%-140%。在温润空气中能缓慢分解 ,游离出醋酸和水杨酸,内服后对胃略有刺激。到目前为止 ,已应用百余年,近年来又发现了很多新用途,引起了人们的广泛关注。阿司匹林为传统的解热镇痛抗炎药,包括阿司匹林钙脲、阿司匹林锌 、赖氨酸阿司匹林 ,其剂型有片剂 、水溶片、肠溶片、缓释片、泡腾片、胶囊剂、软膏剂、栓剂 、散剂等。在实验中,采用湿法制粒压片工制备阿司匹林片。此工艺制备阿司匹林片有较强的可操作性和质量可靠性。
实验目的:通过实验掌握湿法制粒的工艺及含量测定方法,并对该处方、工艺进行评价。方法:实验制备并进行含量测定。
[ 关键词 ]阿司匹林;湿法制粒;含量测定
1 仪器和药品
1.1 仪器:分析天平,酸碱滴定管,烘箱,电炉,药筛(80目、120
目),尼龙筛(14目、16目),搪瓷盘,乳钵等。
1.2 药品 阿司匹林 (药用 ),淀粉 (药用 ),酒石酸 (药用 ), 0.5mol/lNAOH溶液,0.25mol/l硫酸溶液。
2 阿司匹林制备
2.1 处方 阿司匹林 30g, 淀粉3g,酒石酸 0.15g, 10%淀粉浆 (适量 )。
2.2 制备
2.2.1 10%淀粉浆的制备 将酒石酸细粉溶于少量蒸馏水中,加入淀粉混匀 ,加水至100m1分散均匀,加热制作。
2.2.2 制粒 取阿司匹林细粉与淀粉过80目筛,按 “等量递加法”混合均匀,加10%淀粉浆(50±0.5℃)适量 ,制软材 ,用l8目尼龙筛制粒 ,于60℃鼓风干燥,
3 含量测定
水解后剩余滴定
方法;取本品1.5g,精密称定加氢氧化钠滴定(0.5mol/l)50ml.混合,缓缓煮沸10min,放冷,加酚酞指示液,用硫酸滴定液(0.25mol/l)滴定剩余的氢氧化钠。
由反应得知,硫酸标准也与阿司匹林的摩尔比为1:1.由此可计算阿司匹林含量。
4含量测定结果
滴定消耗硫酸滴定液19.22ml,经计算最后测得阿司匹林含量约为57.7%符合要求。
5 讨论
阿司匹林的湿粒遇铁器常使颗粒微带淡红 色,故应尽量避免与铁器接触,制粒时宜使用尼龙筛。加入酸性辅料酒石酸和枸橼酸(0.3%--0.5%) 以防止原料与铁器接触后变色,亦可延缓水解反应。
湿法制粒时混合的强度和时间对制成颗粒 的硬度以及片剂的硬度和崩解时间都有影响。制 粒前要将物料粉碎成粒度大小相近,粒度均匀的 物料容易混合均匀,过筛将物料分级有利于提高 质量,压片时物料的粒度分布对片剂的硬度、片 重差异、裂片等均有明显影响。实验过程中采用的粉碎方法和混合时间及强度较为恰当。
合理选用粘合剂的种类,对药片质量有较大 影响。粘合剂与润滑剂选用是否恰当,不仅影响制剂的成和外观 ,也影响其内在质量。既可使片剂不能成型和在包装、运输过程中出现松片、裂片,也可能长时问不崩解、溶解和有效成分不能溶出、生物利用度低。实验中采用滑石粉作为润滑 剂,颗粒流动性较好 ,片重差异小。实验中选用10%淀粉浆作为粘合剂,以温浆 加入,对阿司匹林片硬度和溶出度影响较小,质量检查符号规定。本实验用l8目筛制粒,成品合格,原因可能与粒度有一定关系。
在片剂制备中,处方、生产工艺、操作技术及机械设备等方面的
因素均会影响片剂质量,压片过程中可能出现的问题有:松片、裂片、粘冲、崩解迟缓、片重差异大、变色或色斑、麻点、迭片等,应对所出现问题分析,找出原因,并加以解决。但本次实验没有出现以上问题。实验所选处方、制备工艺是合理的,所制备的阿司匹林片质量检查全部合格,实验效果较好,达到预期目的。
[参考文献 ]
[1] 刘绍飞, 王成军. 阿司匹林湿法制粒压片及质量检查[J]. 大理学院学报, 2005, (01)
[2]郑虎.药物化学[M].第五版,北京:人民卫生出版社 ,2003.183.
[3]王晓燕,崔福德.辅料对阿司匹林片剂稳定性的影响[J].中国医药工业杂志,2002.12(3):593-594.
[4].中国新药杂志,2003.12(1):45-46.
[5]顾学裘.药物制剂注解[M].北京:人民卫生出版社 .1983.563.
[6]中国药典委员会.中华人民共和国药典-2000~[M].北京:化学工业出版社。2001.附录:5、79、72、75.
[7]胡熙明,张立平.中国药物大全一西药卷[M].北京: 人民卫生出版社 ,1991.62.
[8]罗明生,高天惠.药剂辅料大全[M].四川:四川科技文献 出版社 ,1993.72.
第二篇:实验三 阿司匹林肠溶片的含量测定
实验三 阿司匹林肠溶片的含量测定
一、目的
1、掌握两步滴定法测定阿司匹林含量的原理和方法。
2、掌握剩余滴定法的一般方法和计算。
二、实验内容
(一)阿司匹林肠溶片的含量测定
取本品10片,研细,用中性乙醇70ml,分数次研磨,并移入100ml量瓶中,充分振摇,再用水适量洗涤研钵数次,洗液合并于量瓶中,再用水稀释至刻度,摇匀,滤过,精密量取滤液10ml(相当于阿司匹林0.3g),置锥形瓶中,加中性乙醇(对酚酞指示液显中性)20ml,酚酞指示液3滴,滴加氢氧化钠滴定液(0.1mol/L)至溶液显粉红色,再精密加氢氧化钠滴定液(0.1mol/L)40ml,置水浴上加热15分钟,并时时振摇,迅速放冷至室温,用硫酸滴定液(0.05mol/L)滴定,并将滴定结果用空白试验校正。每1ml的氢氧化钠滴定液(0.1mol/L)相当于18.02mg的C9H8O4。
本品含阿司匹林应为标示量的95.0~105.0%。
(二)硫酸滴定液(0.05mol/L)的标定
取在270~300℃干燥至恒重的基准无水碳酸钠约0.15g,精密称定,加水50ml使溶解,加甲基红-溴甲酚绿混合指示液10滴,用本液滴至溶液由绿色转变为紫红色时,煮沸2分钟,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1ml的硫酸滴定液(0.05mol/L)相当于5.30mg的无水碳酸钠。根据本液的消耗量及无水碳酸钠的取用量,算出本液的浓度,即得。
三、说明
1、为消除阿司匹林的水解产物水杨酸、醋酸及稳定剂枸椽酸、酒石酸对测定的影响,中国药典采用两步滴定法测定本品的含量。
2、中性乙醇的制备方法为:取乙醇,加酚酞指示液适量,滴加氢氧化钠液至显粉红色,即得。
3、过滤供试液,是为了滤除不溶解的附加剂,以免对测定造成影响。为了保证过滤前后供试液的浓度相等,应用干燥滤纸过滤,并弃去初滤液,取续滤液备用。
4、标定硫酸滴定液时,由于在近终点,滴定溶液中存在缓冲对H2CO3和 ,可使终点不敏锐,所以需加热煮沸2分钟,除去其中的H2CO3,再迅速放冷至室温,继续滴定至终点。
,可使终点不敏锐,所以需加热煮沸2分钟,除去其中的H2CO3,再迅速放冷至室温,继续滴定至终点。
四、思考题
1、试简述两步滴定法测定阿司匹林含量的基本原理。
2、测定阿司匹林含量时为什么要做空白试验?应如何做空白试验?
3、为什么要用中性乙醇溶解样品?如何制备中性乙醇?
4、如何理解“取基准无水碳酸钠约0.15g,精密称定”这句话?
实验一 氯化钠的杂质检查
一、目的
1、了解药物中杂质检查的意义。
2、掌握氯化钠中杂质检查的原理和方法。
3、掌握杂质限量的计算方法
二、实验内容
(一)标准溶液的配制
1、标准氯化钠溶液的制备
称取氯化钠0.165g,置1000ml量瓶中,加水适量使溶解并稀释至刻度,摇匀,作为贮备液。
临用前,精密量取贮备液10ml置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10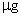 的Cl)。
的Cl)。
2、标准硫酸钾溶液的制备
称取硫酸钾0.181g,置1000ml量瓶中,加水适量使溶解并稀释至刻度,摇匀,即得(每1ml相当于100 的
的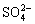 )。
)。
3、标准铁溶液的制备
称取硫酸铁铵[FeNH4(SO4)2·12H2O]0.863g,置1000ml量瓶中,加水溶解后,加硫酸2.5ml,用水稀释至刻度,摇匀,作为贮备液。
临用前,精密量取贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10的Fe3+)。
4、标准铅溶液的制备
称取硝酸铅0.160g,置1000ml量瓶中,加硝酸5ml与水50ml溶解后,用水稀释至刻度,摇匀,作为贮备液。
临用前精密量取贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10的Pb2+)。
配制与贮存用的玻璃容器均不得含有铅。
5、标准砷溶液的制备匀,即得(每1ml相当于1的As3+)。
(二)检查方法
1、酸碱度
取本品5.0g,加水50ml溶解后,加溴麝香草酚蓝指示液2滴,如显黄色,加氢氧化钠液(0.02mol/L)0.10ml,应变为蓝色;如显蓝色或绿色,加盐酸液(0.02mol/L)0.2ml,应变为黄色。
2、溶液的澄清度
取本品5.0g,加水25ml溶解后,溶液应澄清。
3、碘化物
取本品的细粉5.0g,置瓷蒸发皿内,滴加新配制的淀粉混合液[取可溶性淀粉0.25g,加水2ml,搅匀,再加沸水至25ml,随加随搅拌,放冷,加硫酸液(0.025mol/L)2ml,亚硝酸钠试液3滴与水25ml,混匀]适量使晶粉湿润,置日光下(或日光灯下)观察,5分钟内晶粒不得显蓝色痕迹。
4、溴化物
取本品2.0g,加水10ml使溶解,加盐酸3滴与氯仿1ml,边振摇边滴加2%氯胺T溶液(临用新制)3滴,氯仿层如显色,与标准溴化钾溶液(精密称取在105℃干燥至恒重的溴化钾0.1485g,加水使溶解成100ml,摇匀)1.0ml用同一方法制成的对照液比较,不得更深。
5、硫酸盐
取本品5.0g,加水溶解成约40ml(溶解如显碱性,可滴加盐酸使成中性),溶液如不澄清,应滤过,置50ml纳氏比色管中,加稀盐酸2ml,摇匀,即得供试溶液。另取标准硫酸钾溶液1.0ml置50ml纳氏比色管中,加水使成约40ml,加稀盐酸2ml,摇匀,即得对照溶液。于供试溶液与对照溶液中,分别加入25%氯化钡溶液5ml,用水稀释使成50ml,充分摇匀,放置10分钟,同置黑色背景上,从比色管上方向下观察,比较,供试品管不得更浓(0.002%)。
6、钡盐
取本品4.0g,加水20ml,溶解后,滤过,滤液分为两等份,1份中加稀硫酸2ml,另一份中加水2ml,静置15分钟,两液应同样澄清。
7、钙盐
取本品2.0g,加水10ml使溶解,加氨试液1ml,摇匀,加草酸铵试液1ml,5分钟内不得发生浑浊。
8、镁盐
取本品1.0g,加水20ml使溶解,加氢氧化钠试液2.5ml与0.05%太坦黄溶液0.5ml,摇匀,产生的颜色与标准镁溶液(精密称取在800℃炽灼至恒重的氧化镁16.58mg,加盐
称取三氧化二砷0.132g,置1000ml量瓶中,加20%氢氧化钠溶液5ml溶解后,用适量的稀硫酸中和,再加稀硫酸10ml,用水稀释至刻度,摇匀,作为贮备液。
临用前,精密量取贮备液1ml,置100ml量瓶中,加稀硫酸1ml,用水稀释至刻度,摇酸2.5ml与水适量使溶解成1000ml,摇匀)1.0ml,用同一方法制成的对照液比较,不得更深。(0.001%)。
9、钾盐
取本品5.0g,加水20ml溶解后,加稀醋酸2滴,加四苯硼钠溶液(取四苯硼钠1.5g,置乳钵中,加水10ml研磨后,再加水40ml,研匀,用质密的滤纸滤过,即得)2ml,加水使成50ml,如显浑浊,与标准硫酸钾溶液12.3ml用同一方法制成的对照液比较,不得更浓(0.02%)。
10、干燥失重
取供试品,混合均匀(如为较大的结晶,应先迅速捣碎,使成2mm以下的小粒)。分取约1g,平铺在130℃干燥至恒重的扁形称瓶中,厚度不超过5mm,精密称定。将瓶盖取下,置称瓶旁,或将瓶盖半开,置烘箱内于130℃干燥约2~3小时后,将称瓶盖好,取出,置干燥器中放置40分钟后,称定重量。再按上述方法自“将瓶盖取下……”起,继续干燥1~2小时,依法操作,至恒重为止。从减失重量和取样量计算供试品的干燥失重(规定不得超过0.5%)。
11、铁盐
取本品5.0g,加水溶解成25ml,置50ml纳氏比色管中,加稀盐酸4ml与过硫酸铵50mg,用水稀释成35ml后,加30%硫氰酸铵溶液3ml ,再加水适量稀释成50ml,摇匀。如显色,立即与标准铁溶液1.5ml用同一方法制成的对照液比较,不得更深(0.0003%)。
12、重金属
取本品5.0g,加水20ml溶解后,置25ml纳氏比色管中,加醋酸盐缓冲液(pH3.5)2ml与水适量使成25ml,加硫代乙酰胺试液2ml,摇匀,放置2分钟,置白纸上,自上向下透视。如显色,立即与标准铅溶液1.0ml用同一方法制成的对照液比较,不得更深(0.0002%)。
13、砷盐
仪器装置,如图。A为100ml标准磨口锥形瓶,B为中空的标准磨口塞,上连导气管C(外径8.0mm,内径6.0mm),全长约180mm。D为具孔的有机玻璃旋塞,其上部为圆形平面,中央有一圆孔,孔径与导气管C的内径一致,其下部孔径与导气管C的外径相适应。将导气管C的顶端导入旋塞下部孔内,并使管壁与旋塞的圆孔适相吻合,粘合固定,E为中央具有圆孔(孔径6.0mm)的有机玻璃旋塞盖,与D紧密吻合。

测试时,于导气管C中装入醋酸铅棉花60mg(装管高度约60~80mm),再于旋塞D的顶端平面上放一片溴化汞试纸(试纸大小以能覆盖孔径而不露出平面外为宜),盖上旋塞E并旋紧,即得。
标准砷斑的制备:精密量取标准砷溶液2ml,置A瓶中,加盐酸5ml与水21ml,再加碘化钾试液5ml与酸性氯化亚锡试液5滴,在室温放置10分钟后,加锌粒2g,立即照上法装妥的导气管C密塞于A瓶上,并将A瓶置25~40℃水浴中,反应45分钟后,取出溴化汞试纸,即得。
检查法,取本品5.0g,置A瓶中,加水23ml溶解后,加盐酸5ml,照标准砷斑的制备,自“加碘化钾试液5ml”起,依法操作,将生成的砷斑与标准砷斑比较,不得更深(0.00004%)。
三、说明
1、药物杂质检查必须严格遵守平行原则。平行原则是指样品与标准必须在同一条件下进行反应与比较。即应选择容积、口径和色泽相同的比色管,在同一光源、同一衬底上,以相同的方式(一般是自上而下)观察,加入的试药种类、量,加入顺序和反应时间等也应一致。
2、杂质限量是指药物中杂质的最大允许量。其计算公式为:

3、药物的杂质检查一般为限量检查,合格者仅说明其杂质量在药品质量标准允许范围内,并不说明药品中不含该项杂质。
四、思考题
1、药物中杂质检查的意义是什么?
2、药物中杂质的来源主要有哪些?什么是一般杂质?什么是特殊杂质?
3、药物中杂质检查应严格遵循什么原则?为什么?
4、试计算出氯化钠中溴化物、硫酸盐、镁盐、钾盐、铁盐、重金属和砷盐的限量。
5、取某一药物0.5g进行重金属检查,药典规定限量为10ppm,应取多少ml标准铅溶液?
6、药典规定某一药物砷盐限量为4ppm,取标准砷溶液2ml作对照,问应取供试品多少克
实验三 阿司匹林肠溶片的含量测定
一、目的
1、掌握两步滴定法测定阿司匹林含量的原理和方法。
2、掌握剩余滴定法的一般方法和计算。
二、实验内容
(一)阿司匹林肠溶片的含量测定
取本品10片,研细,用中性乙醇70ml,分数次研磨,并移入100ml量瓶中,充分振摇,再用水适量洗涤研钵数次,洗液合并于量瓶中,再用水稀释至刻度,摇匀,滤过,精密量取滤液10ml(相当于阿司匹林0.3g),置锥形瓶中,加中性乙醇(对酚酞指示液显中性)20ml,酚酞指示液3滴,滴加氢氧化钠滴定液(0.1mol/L)至溶液显粉红色,再精密加氢氧化钠滴定液(0.1mol/L)40ml,置水浴上加热15分钟,并时时振摇,迅速放冷至室温,用硫酸滴定液(0.05mol/L)滴定,并将滴定结果用空白试验校正。每1ml的氢氧化钠滴定液(0.1mol/L)相当于18.02mg的C9H8O4。
本品含阿司匹林应为标示量的95.0~105.0%。
(二)硫酸滴定液(0.05mol/L)的标定
取在270~300℃干燥至恒重的基准无水碳酸钠约0.15g,精密称定,加水50ml使溶解,加甲基红-溴甲酚绿混合指示液10滴,用本液滴至溶液由绿色转变为紫红色时,煮沸2分钟,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1ml的硫酸滴定液(0.05mol/L)相当于5.30mg的无水碳酸钠。根据本液的消耗量及无水碳酸钠的取用量,算出本液的浓度,即得。
三、说明
1、为消除阿司匹林的水解产物水杨酸、醋酸及稳定剂枸椽酸、酒石酸对测定的影响,中国药典采用两步滴定法测定本品的含量。
2、中性乙醇的制备方法为:取乙醇,加酚酞指示液适量,滴加氢氧化钠液至显粉红色,即得。
3、过滤供试液,是为了滤除不溶解的附加剂,以免对测定造成影响。为了保证过滤前后供试液的浓度相等,应用干燥滤纸过滤,并弃去初滤液,取续滤液备用。
4、标定硫酸滴定液时,由于在近终点,滴定溶液中存在缓冲对H2CO3和,可使终点不敏锐,所以需加热煮沸2分钟,除去其中的H2CO3,再迅速放冷至室温,继续滴定至终点。
四、思考题
1、试简述两步滴定法测定阿司匹林含量的基本原理。
2、测定阿司匹林含量时为什么要做空白试验?应如何做空白试验?
3、为什么要用中性乙醇溶解样品?如何制备中性乙醇?
4、如何理解“取基准无水碳酸钠约0.15g,精密称定”这句话?
实验四 复方磺胺甲恶唑片的含量测定
一、目的
1、掌握复方制剂的分析特点及赋形剂的干扰与排除方法。
2、掌握双波长分光光度法测定复方磺胺甲口恶 唑片中磺胺甲口恶 唑与甲氧苄啶含量的原理与方法。
二、实验内容
1、供试品溶液的制备
取本品10片,精密称定,研细,精密称取适量(约相当于磺胺甲口恶 唑50mg与甲氧苄啶10mg),置于100ml量瓶中,加乙醇适量,振摇15分钟磺胺甲口恶 唑与甲氧苄啶溶解,加乙醇稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
2、对照品溶液的制备
精密称取在105℃干燥至恒重的磺胺甲口恶 唑对照品50mg与甲氧苄啶对照品10mg,分置100ml量瓶中,各加乙醇溶解并稀释至刻度,摇匀,分别作为对照品溶液(1)与对照品溶液(2)。
3、磺胺甲口恶 唑的测定
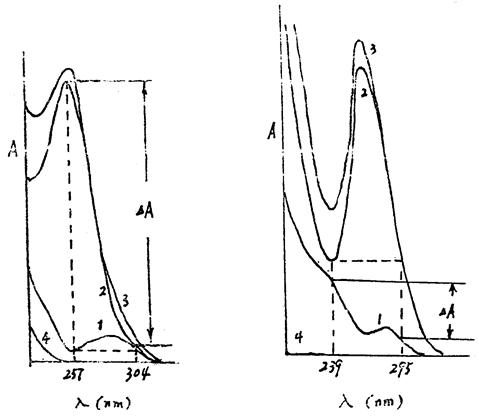
精密量取供试品溶液与对照品溶液(1)、(2)各2ml,分置100ml量瓶中,各加0.4%氢氧化钠溶液稀释至刻度,摇匀。取对照品溶液(2)的稀释液,以257nm为测定波长( 2),在304nm波长附近(每间隔0.5nm)选择等吸收点波长为参比波长(1),要求
2),在304nm波长附近(每间隔0.5nm)选择等吸收点波长为参比波长(1),要求 A=A2-A1=0。再在2与1波长处分别测定供试品溶液的稀释液与对照品溶液(1)的稀释液的吸收度,求出各自的吸收度差值(A)计算,即得。
A=A2-A1=0。再在2与1波长处分别测定供试品溶液的稀释液与对照品溶液(1)的稀释液的吸收度,求出各自的吸收度差值(A)计算,即得。
4、甲氧苄啶的测定
精密量取上述供试品溶液与对照品溶液(1)(2)各5ml,分置100ml量瓶中,各加盐酸-氯化钾溶液[取盐酸液(0.1mol/L)75ml与氯化钾6.9g,加水至1000ml]摇匀稀释至刻度,摇匀。取对照品溶液(1)的稀释液,以239.0nm为测定波长(2),在295nm波长附近(每间隔0.2nm)选择等级吸收点波为参比波长(1),要求A=A2-A1=0。再在2与1波长处分别测定供试品溶液的稀释液与对照品溶液(2)的稀释液的吸收度,求出各自的吸收度差值(A),计算,即得。
本品每片中含磺胺甲口恶 唑(C10H11N3O3S)应为0.360~0.440g,含甲氧苄啶(C14H12N4O3)应为72.0~88.0mg。
(三)说明
1、 磺胺甲口恶 唑与甲氧苄啶的结构分别为:
 磺胺甲口恶唑 (SMZ)
磺胺甲口恶唑 (SMZ)
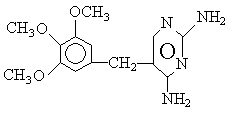 甲氧苄啶 (TMP)
甲氧苄啶 (TMP)
SMZ与TMP的紫外吸收图谱分别为:

2、复方磺胺甲口恶 唑片的处方为:
磺胺甲口恶 唑 400g
甲氧苄啶 80g 制成1000片
3、本实验采用双波长分光光度法测定SMZ和TMP的含量。由于干扰组分在测定波长和参比波长处吸收度相等,可在计算A时消去,所以应用本法可以不经分离,直接测定复方制剂中SMZ和TMP的含量。
4、本品在中国药典(1985年版)中的名称为复方磺胺甲基异口恶 唑片,其含量测定方法为:磺胺甲基异口恶 唑以重氮化法测定;甲氧苄啶以氯仿提取后,用紫外分光光法测定。
5、此实验的原理可参考:胡育筑,等。南京药学院学报,1982,(1):29-35。
四、思考题
1、试简述双波长分光光度法的原理。
2、试比较中国药典(1985年版)与中国药典(20##年版)中本品含量测定方法的优缺点。
3、试查阅本品的其它分析方法,从中了解复方制剂分析的特点及发展趋势。
-
阿司匹林的合成实验报告
阿司匹林的合成高分子113班09一实验原理阿司匹林为解镇痛药用于治疗伤风感冒头痛发烧神经痛关节痛及风湿病等近年来又证明它具有抑制血…
- 阿司匹林的制备实验报告
- 阿司匹林的制备实验报告
-
阿司匹林实验报告
综合实验报告题目阿司匹林的合成表征及含量测定班徳忠姓专业班级工业分析2学号0806060213同组者蔡召志指导教师万其进喻20xx…
-
实验报告 阿司匹林的合成
阿司匹林的合成一实验目的通过阿司匹林的合成掌握酯化反应和精制原理及基本操作熟悉药物合成实验装置的安装和使用掌握水杨酸的限量检查方法…
-
实验七 阿司匹林片的制备
试验七片剂第一部分片剂制备与部分质量检查一实验目的1通过阿司匹林片剂制备掌握湿法制粒压片的工艺过程2了解单冲与11冲压片机的基本构…
-
有关阿司匹林制法的开放实验报告
开放实验总结报告学生姓名班级学号所在院系开放实验室名称日期北京理工大学实验室设备处制一实验项目概况二实验项目技术报告于氢氧化碱溶液…
-
阿司匹林的合成实验报告
阿司匹林的合成高分子113班09一实验原理阿司匹林为解镇痛药用于治疗伤风感冒头痛发烧神经痛关节痛及风湿病等近年来又证明它具有抑制血…
-
阿司匹林20xx(实验室版实验报告)
阿司匹林合成论文阿司匹林合成摘要通过该项实验可以使同学们掌握有机物质分离提纯的方法了解乙酰水杨酸的化学性质乙酰水杨酸俗名阿司匹林又…
- 阿司匹林的制备实验报告
- 阿司匹林的制备实验报告