BV临床试验总结报告
体外诊断试剂临床试验
总结报告
产品通用名:细菌性阴道病(BV)检测试剂盒(唾液酸酶法)
型号规格:50人份/盒
研究开始日期:20xx年07月29日
研究完成日期:20xx年10月31日
研究单位:中国人民解放军第八五医院
蚌埠医学院第二附属医院
临床试验类别:“已有同品种批准上市”产品的临床研究
注册申请人:北京金沃夫生物工程科技有限公司
联系电话:010-60216810
报告日期:20xx年05月23日
目录
研究摘要 试验研究人员 缩略语 1、引言
2、临床试验目的 3、试验管理 4、临床试验设计 5、试验方法 6、试验结果的处理及分析方法7、临床试验结果 8、结论 附件
--------------------------------- --------------------------------- --------------------------------- --------------------------------- --------------------------------- --------------------------------- ---------------------------------- ---------------------------------- ----------------------------------
---------------------------------- ---------------------------------- ----------------------------------
1
2 2 2 3 3 4 4 5 5 6 7 7
研究摘要
目的:依据批准的临床方案对其生产的细菌性阴道病(BV)检测试剂盒(唾液酸酶法)进行临床研究,统计两者检测结果的灵敏度、特异性、符合率等相关指标,以验证其研制产品与已上市同类产品的等效性。
方法:选用珠海迪尔生物工程有限公司的细菌性阴道病(BV)检测试剂盒(唾液酸酶法)进行对比,收集医院妇产科病人阴道分泌物拭子,分别用金沃夫试剂与珠海迪尔试剂进行对比检测,记录检测结果,对检测不一致的样本再用Amsel法进行验证。
结论:经临床验证,用对比试剂和被评价试剂共检测临床阴道拭子234例,被评价试剂的阳性符合率97.10%、阴性符合率97.58%,总符合率为97.44%,与珠海迪尔同类产品具有良好的一致性,具有等效性。因此,该试剂可临床用于对细菌性阴道病的定性检测,辅助诊断细菌性阴道病感染。
试验研究单位
中国人民解放军第八五医院
蚌埠医学院第二附属医院
缩略语
BV:Bacterial Vaginosis,细菌性阴道病
2
1、引言
与临床试验产品有关的背景情况
1.1 目标分析物简介
细菌性阴道病(Bacterial Vaginosis,简称BV )是指由于正常阴道菌群减少,被一组厌氧菌,包括阴道加德纳尔菌(Gardnerella Vaginalis)、类杆菌 (Bacteides spp) ,和普氏杆菌属 (Prevotilla spp)等所取代,而引起的一种临床综合症。19xx年在斯德哥尔摩国际会议正式命名为细菌性阴道病。细菌性阴道病(BV)是妇产科最常见的疾病之一,感染率15-50%,且易复发。BV 引起的直接危害是患者感觉不适,但若不及时和正确治疗,将增加并发症出现的可能。并发症包括输卵管炎、子宫内膜炎、盆腔炎、宫外孕、泌尿感染、术后感染及妇科肿瘤等,临床研究表明,BV 更直接的危害是胎盘感染、胎膜早破、早产、羊水感染、组织性绒毛膜炎、低体重新生儿,新生儿黄胆升高及在剖宫产术后宫内膜炎及其它妊娠不良和妊娠并发症的危险因素,严重威胁母婴健康。
BV检测的金标准是Amsel法,该方法影响因素众多,氨试验不符合生物安全要求,PH无法准确检测,白带稀薄无判断标准,所以不适合推广使用。
近年来的研究证实,细菌性阴道病分泌物内一组厌氧菌显示异常唾液酸酶活性,BV细菌性阴道病快速检测试剂(唾液酸酶法)用于检测阴道分泌物内异常厌氧菌群。
1.2 临床预期使用目的
北京金沃夫生物工程科技有限公司研制开发的细菌性阴道病(BV)检测试剂盒(唾液酸酶法)可用于细菌性阴道病的检测。
1.3 产品方法学、原理
本试剂盒是根据患BV的妇女阴道分泌物显示出异常的唾液酸酶活性这一原理制成,利用一种特殊的唾液酸酶底物与提取后的BV患者阴道分泌物中存在的特异性唾液酸酶作用,可以使该底物迅速水解,加入适当的显色剂后,会发生明显的颜色变化而进行检测。
1.4 国内外批准上市情况和产品应用现状
国内已有同类产品批准注册企业的有珠海迪尔生物工程有限公司、深圳市赛尔生物技术有限等近十家,进口的有美国Gryphus Diagnostics,L.L.C.生产的BV BLUE。目前在北京,上海等各医院都已经普及唾液酸酶法。
2、临床试验目的
对北京金沃夫生物工程科技有限公司开发的细菌性阴道病(BV)检测试剂盒 3
(唾液酸酶法)与已批准上市的珠海迪尔生物工程有限公司同类产品针对临床样本进行对比试验研究,统计两者检测结果的灵敏度、特异性、符合率等相关指标,以验证其研制产品与已上市同类产品的等效性。
3、试验管理
承担本临床试验的中国人民解放军第八五医院、蚌埠医学院第二附属医院,医院均属于省级医院,医院资质符合体外诊断试剂临床试验的要求。
按照《体外诊断试剂临床研究技术指导原则》要求本试验确定了相关的研究人员,建立了相应的质量控制管理制度,确保该试验的科学性和严谨性。
4、临床试验设计
北京金沃夫生物工程科技有限公司研制的细菌性阴道病(BV)检测试剂盒(唾液酸酶法),属于Ⅱ类医疗器械体外诊断试剂。本临床试验方案依据国家食品药品监督管理局《体外诊断试剂注册管理办法(试行)》及《体外诊断试剂临床研究技术指导原则》等相关法令及规定制定。
4.1临床研究对象
本次临床试验共考核临床病例234例,样本均来源于两家医院妇产科科住院或门诊病人。具体分配见下表:
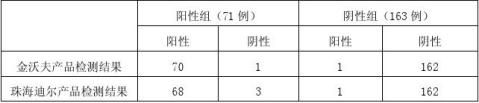
注:阳性组入围组是疑似细菌性阴道病的病例及已确诊细菌性阴道病的病例。
4.2 样本采集、保存方法
本临床试验样品为阴道分泌物拭子,由医院工作人员负责取样。用消毒拭子尽可能多地收集阴道分泌物(注:采样前病人不得在24小时内使用阴道乳状药剂,不得收集宫颈处的阴道分泌物),分成3份。收集后可立即检测,如不能立即检测应2-8℃冷藏保存,但不能超过6个小时。
4.3 试验材料和对比试剂的确立
(1)被评试剂:北京金沃夫生物工程科技有限公司研制的细菌性阴道病(BV)检测试剂盒(唾液酸酶法),产品批号: 20110701 ,规格型号 50人份/盒 , 4
有效期 20xx年1月27日 。
(2)对比试剂:BV细菌性阴道病快速检测试剂盒(唾液酸酶法),厂家: 珠海迪尔生物工程有限公司,注册证号: 粤食药监械(准)字2008第2400320号 。
对比试剂选择标准:选择目前临床普遍认为质量较好的、已批准上市的同品种产品。其方法学应采用唾液酸酶法;其临床适用范围应为定性检测细菌性阴道病。因此,我们选择已批准上市、临床广泛采用且质量较好的珠海迪尔生物工程有限公司生产的同类产品。
(3)Amsel法诊断标准:(1)阴道 PH 值> 4.5 ;(2)阴道分泌物增多,有异味,变稀;(3)阴道分泌物加 10%KOH 可闻到鱼腥味;(4)阴道涂片可见线索细胞( Clue Cell )。其中三项为阳性,即判定为 BV 阳性。
5.试验方法
分别用北京金沃夫生物工程科技有限公司和珠海迪尔生物工程有限公司各自生产的细菌性阴道病检测试剂盒(唾液酸酶法)对样品分别进行对比检测。对检测结果不一致的样本,应采用Amsel法再次进行确认。
操作步骤按照各个厂家提供的产品说明书的要求进行。
6.试验结果的处理及分析方法
试验时,应将试验结果记录在试验原始记录表上。试验原始记录表应包括病人门诊编号、姓名、年龄、试验结果、两种方法不一致试验结果的确认及分析等内容。
试验结束后,由临床研究单位整理试验数据,并对试验数据进行统计分析,依据结果评价考评试剂与对比试剂在临床上用于BV感染诊断的阳性符合率、阴性符合率和总符合率。
本试验的临床评价指标为临床灵敏度、特异性和符合率。
采用评价试验真实性的资料归纳表[方表(Square Table)]进行统计分析。
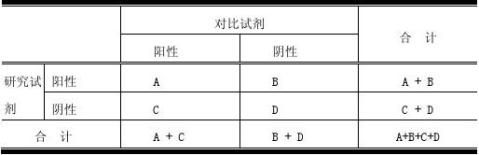
灵敏度(真阳性率)=A/(A+C)×100%
特异度(真阴性率)=D/(B+D)×100%
符合率=(A+D)/(A+B+C+D)×100%
并对上述[方表(Square Table)]资料进行Kappa一致性分析,Kappa系 5
数>0.8,为高度一致,认为两系统等效; 0.4<Kappa系数<0.8认为一致,需进行阳性符合率和阴性符合率比较并进行统计学分析;Kappa系数<0.4则认为两系统不一致,两系统不等效。本试验要求Kappa系数≥0.8。
7.临床试验结果
本次临床试验共考核临床样本234例,两试剂检测共同阳性67例,共同阴性161例。统计结果见表2、3。 表2:阳、阴性组检测结果
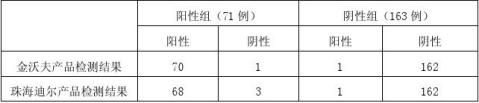
表3:所有检测结果汇总
珠海迪尔产品检测结果
项 目
阳性结果(+)
金沃夫产品 检测结果
阳性结果(+) 阴性结果(-)
67 2 69
阴性结果(-)
4 161 165
71 163 234 合 计
合 计
金沃夫细菌性阴道病(BV)检测试剂盒(唾液酸酶法)和珠海迪尔公司同类产品相比:
阳性符合率=67/(67+2)×100%=97.10% 阴性符合率=161/(161+4)×100%=97.58%
总符合率=(67+161)/(67+2+161+4)×100%=97.44%
将表3的分析结果输入SPSS15统计软件进行KAPPA检验,对两种诊断方法的结果一致性进行分析,KAPPA检验的结果为:KAAPA=0.939,P<0.0001。因此,金沃夫细菌性阴道病(BV)检测试剂盒(唾液酸酶法)和珠海迪尔公司同类产品相比,两种产品检测结果具有显著的一致性。
8.讨论与结论
6
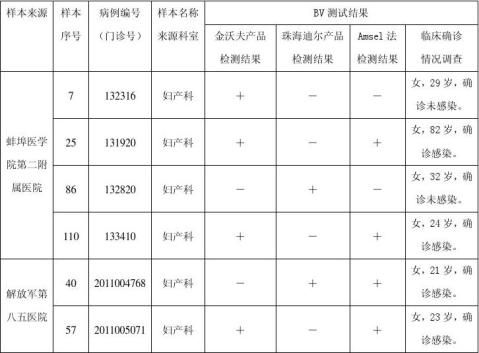
(1)检测结果不一致样本汇总
(2)不一致结果产生的原因分析
对检测不一致的样本的分析:其中,有6份样本检测结果不一致,其中4份金沃夫试剂检测为阳性,珠海迪尔试剂检测为阴性,2份金沃夫试剂检测为阴性,珠海迪尔试剂检测为阳性,这6份不一致的产品经Amsel法再次验证,4份样本为阳性,2份样本为阴性。
针对不一致的结果,我们分析认为其产生原因有以下几点:
1)金沃夫细菌性阴道病(BV)检测试剂盒(唾液酸酶法)及同类产品均为定性产品,存在发生检测结果错误的可能性,因此可能造成同一样本出现不一致的检测结果,但是从预期用途来说,作为临床辅助诊断产品,出现小概率的错误结果,是可以被临床应用所接受的。
2)不同厂家提供的产品,其最低检测限存在一定差异,因此对于低浓度的阳性样本,可能会出现不同产品检测结果不一致的情况。
3)另外,本类定性产品在检测阳性样本时显蓝色,低浓度阳性样本显绿色,阴性样本显示为黄色。根据经验,当阳性样本浓度很低时,绿色结果不是很明显, 7
可能会造成检测人员判读结果出现错误,因此检测人员主观判断也可能造成不一致结果的产生。
4)根据厂家提供的产品说明书以及相关的原理,可能是不同厂家使用的显色剂或者比例不同,同样会造成部分结果的总体上有少量不一致。
但是临床试验表明,金沃夫产品和对照试剂珠海迪尔产品在临床检测的总体结果上有高度的一致性。
北京金沃夫生物工程科技有限公司生产的细菌性阴道病(BV)检测试剂盒(唾液酸酶法)与珠海迪尔生物工程有限公司生产的同类产品对比临床使用研究,两种试剂对细菌性阴道病抗体检测结果的阳性符合率97.10%、阴性符合率97.58%,总符合率为97.44%,两者在临床应用上具有非常好的一致性。
经临床验证,北京金沃夫生物工程科技有限公司生产的细菌性阴道病(BV)检测试剂盒(唾液酸酶法)可临床用于细菌性阴道病的定性检测,用以辅助诊断细菌性阴道病感染。
北京金沃夫生物工程科技有限公司
20xx年05月23日
8
第二篇:I期临床试验总结报告样本
SFDA临床试验批准号:2002XXXXXX号
中国CRO网 收集整理
XXX注射液
Ⅰ期临床人体耐受性试验总结报告
试验单位:XXXXXXXX医院
试验负责:XXX
试验设计:XXX,XXXX
试验日期:20xx年X月X日至X月X日
申办单位:XXXXX有限公司
(以下仅提供报告格式,其数据和文字均为虚构,如有巧合,纯属偶然;数据前后有矛盾之处,也请原谅)
XXX注射Ⅰ期临床人体耐受性试验总结报告
XXX注射Ⅰ期临床人体耐受性试验总结报告
摘 要
单次给药的耐受性试验:30名健康受试者,根据体重和性别随机分配到7个剂量组(组1、组2、组3、组4、组5、组6、组7)。组1、组2每组2人;组3、组4、组5每组6人;组6、组7每组 4人。参照费氏递增法(改良Fibonacci法)递增给药,各组分别静脉滴注XXX注射液5、10、20、30、40、50、60ml,滴速20-30滴/min。每个受试者只接受一个相应的剂量,从小剂量开始,每个剂量应用后未见不良反应,才可用下一个剂量。观察给药后健康人体对XXX注射液的反应和耐受性。
多次(累积)给药的耐受性试验:12名健康受试者根据体重和性别随机分配到甲、乙两个剂量组,每组6人。分别静脉滴注XXX注射液20、10 ml,滴速20-30滴/min,连续5d。 结论:(1)每次按10 ml/次给药,有可能引血胆红素轻度增高或尿胆原阳性,但受试者无明显不适,因此推荐II期临床试验为10 ml/次;15 ml/次有可能引起血胆红素轻度增高或尿胆原阳性,但风险不会太大,可在严密监护下尝试。20 ml/次虽然引起的不良反应尚属轻度,但发生的频度较高,慎用。(2)10 ml/次就有可能引起轻度不良反应,主要为血胆红素轻度增高或尿胆原阳性,但受试者无自觉症状。20 ml/次可引起的不良反应有所增加。这种血胆红素轻度增高或尿胆原阳性,且两次重复结果相近,推测是药物引起的轻度溶血所致。其他主要不良反应为口干(2例)、头晕(1例)、皮疹(1例)。
根据国家药品监督管理局XXX号批文的要求,按照《新药审批办法》,《药品临床试验管理规范》(GCP),《中药新药临床研究的技术要求》,以及《中药新药临床研究指导原则》和XXX的化学组成、功能主治、药效学、毒理学研究资料,于20xx年7月17日~20xx年9月20日,对XXX有限公司申报的XXX注射液中药二类新药,进行Ⅰ期临床人体耐受性试验,总结报告如下。
1 研究目的
选择健康人为受试者,从安全的初始剂量开始,观察人体对XXX注射液的耐受性,为制定本品的Ⅱ期临床试验给药方案提供依据。
2 临床资料与研究方法
2.1 病例选择
2.1.1 入选标准
1.健康志愿者。
2.年龄在 18~50岁,男女各半。
3.体重在标准体重的±10%范围内 [标准体重(kg)=0.7×(身高cm-80)]。
4.无烟酒嗜好。
5.心、肝、肾、血液等检查指标均在正常范围。
6.知情同意,志愿受试。
1
XXX注射Ⅰ期临床人体耐受性试验总结报告
2.1.2 排除标准
1.
2.
3.
4.
5.
6.
7.
8.
2.1.3 剔除标准
对已被选入本临床研究,属于以下情况之一者,作为剔除病例。
1.不符合纳入标准,或符合排除标准者
2.一次药未用者
3.无任何记录者
2.1.4 脱落(退出)标准
对已被选入本临床研究,属于以下情况之一者,作为脱落病例。
1.受试者依从性差,不能按时按量用药。
2.使用其他影响耐受性判断的药物。
3.受试者不愿意继续进行临床试验,向主管医生提出退出者。
2.1.5终止试验标准
1.在剂量递增过程中出现了严重不良反应,虽未达到最大剂量,亦应终止试验。
2.如半数受试者出现轻度不良反应,应终止试验。
3. 在达到最大剂量时,虽未出现不良反应,亦应终止试验。
2.2 研究药物与方法
2.2.1 研究药物
试验药品:XXX注射液,由XXXX有限公司提供,规格:20ml/支。批号:XX。经XXX药品检验所检验,试验用药须符合临床研究用质量标准(草案),批号需与检验批号一致。
2.2.2 分组与给药方法
Ⅰ、单次给药耐受性试验
1.初试剂量确定:根据改良Blach well法计算,小鼠iv和iv的LD50未测出,至少大于210 g/kg生药;犬长毒试验静注90天出现毒性的剂量为17.4 g/kg生药,其 1/60 为0.29 g/kg生药,按成人60 kg计,预计初试剂量为17.4 g生药,相当于注射液6.69 ml;大鼠长毒试验出现毒性剂量为41.6 g/kg, 其1/60 约为0.693 g/kg生药,其按成人60 kg 计,预计初试剂量为41.6 g生药,相当于注射液16 ml。根据以上计算结果,宜选用较小剂量,同时结合临床经验及可操作性,确定本试验的初试剂量为5 ml /日/人。
2 妊娠期、哺乳期妇女。 具有原发性心、肝、肾、血液学疾病者。 精神或法律上的残疾患者。 有酒精、药物滥用病史。 过敏体质(对两种或两种以上药物或食物、花粉过敏史者)。 3个月内参加过其他药物临床试验者。 正在应用其他预防和治疗的药物者。 研究者认为不能入组的其他原因。
XXX注射Ⅰ期临床人体耐受性试验总结报告
2.最大剂量确定:根据犬长毒试验最小有毒量17.4 g/kg的 1/10计算,成人60 kg的最大剂量为104.4 g,相当于注射液40.15 ml。根据大鼠长毒试验观察到不良反应的最小剂量(5.2 g/kg)的 1/10计算,成人60 kg的最大剂量为156 g,相当于注射液60 ml。结合临床可操作性,确定本试验的最大剂量为60 ml /日/人。
3.剂量递增方案:见表1,参照费氏递增法(改良Fibonacci法)递增。每个受试者只接受一个相应的剂量。从小剂量开始,每个剂量应用后未见不良反应,才可用下一剂量出现较重不良反应时或如半数受试者出现轻度不良反应,即使未达到最大剂量,均应停止试验。 表1 剂量递增方案
1 组别
-- 递增比例
5 用量/ml
2 预设人数
每组男女各半
2 +100.0% 10 4
3 +100.0% 20 6
4 +50.0% 30 6
5 +33.3% 40 6
6 +25% 50 6
7 +20% 60 6
4.试验例数:30例,男女各半。
5.分组方法:将男女受试者各15例按体重排序,借助DAS统计分析系统产生随机号,产生受试者序号,按序入组。试验从第1组开始,参看表2。 表2 根据体重随机分组表 受试者序号
9 2 12 13 10 5 15 4 7 11 6 14 3 1 8
Ⅱ、累积性耐受性试验
1. 剂量:根据单次用药试验结果,预做2个剂量组。申办者已知本药有效剂量范围为10-20ml,为追求安全性,建议累积性试验剂量限于此剂量范围。
2.疗程:每日一次,连续静脉滴注5天。 3. 受试者例数:男女各6 人。
3
男性受试者
组别 体重/kg
最重 5
次重 2 6
7 6 4 7 4 5 6 4 7 3 1 5
-- -- -- -- -- -- -- -- -- -- -- -- 最轻
剂量/ml 35 10 45 60 45 25 60 25 35 45 25 60 15 5 35
受试者序号
10
9 14 5 2 6 13 3 15 12 8 7 11 1 4
女性受试者 组别 体重/kg
最重 6
次重 5 7
4 2 4 7 3 7 6 5 5 6 1 4
-- -- -- -- -- -- -- -- -- -- -- -- 最轻
剂量/ml 45 35 60 25 10 25 60 15 60 45 35 35 45 5 25
XXX注射Ⅰ期临床人体耐受性试验总结报告
Ⅲ、给药方法
均将相应剂量的药物加于5%葡萄糖注射液250 ml 内静脉滴注。(有糖尿病或病史者可改用生理盐水250 ml),每日一次。单次给药剂量组共1日,连续给药剂量组共5日。
2.2.3观察项目
1. 人口学特征:性别,年龄,身高,体重,民族,职业。
2. 一般情况:观察试验前和试验后不同时间点心率、心律、呼吸、血压、体温。
3. 实验室检查:观察试验前后不同时间血常规、出血时间、凝血时间、尿常规(8项)、大
便常规(加潜血)、血液生化(包括肝功能TBIL、DBIL、ALT、TP、ALB、GGT、肾功能BUN、Cr、血脂TG、TC、HDL、LDL、血GLU)、血糖、ECG。
4. 其他检查:观察试验前进行乙肝全项、胸透、B超(肝、胆、脾、胰、肾)检查以筛选。
5. 不良反应观察(见后)。
所有实验室检查结果如出现异常,则重复测定一次,以进一步证实。
2.2.4观察时点
1. 受试者筛选:试验当日8:00AM前完成受试病例登记、体检、理化检查等。
2. 准备期:试验前禁食 12 h。
3. 试验期:8:00AM开始静滴。给药后1、2、4、6、8、12、24h观察记录一般情况、临床症状和体征,第24小时进行各项理化检查。如果出现不良反应,随时作相应的理化检查。试验期间,每次只做一个剂量组。试验从初试剂量至高剂量逐个剂量组依次进行。不可同时进行2 个以上剂量组的试验。
4. 随访期:无不良反应者,停药后随访3天,观察有无不良反应出现,同时观察一般情况、临床症状和体征,进行各项理化检查。对实验中出现不良反应者,应随防至症状或体征及相应理化检查恢复正常。
与药物与不良事件间相关性分析,根据肯定有关、可能有关、难以判断、可能无关、肯定无关进行逐一判断。肯定相关和可能相关视为不良反应。实验室和其他检查中,如出现无临床意义的改变不视作不良反应。
2.3 不良事件记录、质量控制与伦理学要求
严格按GCP,以及相关法规进行,具体细节见研究方案。
2.4 数据管理和分析
1. 数据的采集:①研究者必须保证数据真实、完整、准确。②研究记录所有项目均需填写,不得空项、漏项(无记录的空格划斜线);做任何更正时只能划线,旁注改后的数据,说明理由,由研究者签名并注明日期,不得擦涂、覆盖原始记录。③实验室检查项目齐全。试验病例完成观察后3天内将研究记录等资料交研究负责人审核,10天内将研究病历交项目负责人。
2. 数据的监查:监察员审核每份原始研究记录表,并逐份填写“监察员审核页”,确认临床试验数据记录及时、准确、规范、完整。监查员每次访视后书写“临床试验监查报告”。 4
XXX注射Ⅰ期临床人体耐受性试验总结报告
3. 数据检查和录入:由统计单位数据管理员校对录入,如有疑问,填写疑问表返回监查员,由研究者对疑问表中的问题进行书面解答并签名,交回数据管理员,输入数据库。疑问表应妥善保管。
4.统计分析:由统计人员完成,内容包括:①由于受试人数较少,个例的结果应结合专业分析。采用DAS软件进行分析。②统计受试者入选数量,脱落和剔除病例情况,人口统计学和其他基线特征及安全性分析。③描述性统计分析,定性指标以频数表,百分率或构成比描述;定量指标以均数,标准差,或最大值、最小值、中位数描述。完成统计后提交统计报告书。
3 试验结果
3.1单次给药的耐受性试验 3.1.1受试者分配
受试者根据体重和性别分配至7个剂量组,每组人数及给药剂量见表3。试验期间无退出和剔除者。
表3 受试者分组 组1
5 用药剂量/ml
2 参加人数
0 退出人数
0 剔除人数
3.1.2 各组受试者一般资料
各组性别相当,年龄、身高、体重、民族、职业等资料相近,无重要的既往病史和药物过敏史(见表4)。
表4 各组受试者一般资料 组1 n 2
1 性别 男
1 女
民族
6 汉族 0 非汉族
职业
3 体力 3 非体力
年龄(岁)
mean 22.5 SD 3.5 25 最大值 20 最小值 22.5 中位数
身高(cm)
组2
10 2 0 0 组3 20 6 0 0 组4 30 6 0 0 组5 40 6 0 0 组6 50 4 0 0 组7 60 4 0 0
组2 2 1 1 6 0 2 4 24.0 2.8 26 22 24.0 组3 6 3 3 6 0 4 2 21.0 3.2 25 16 21.5
5
组4 6 3 3 6 0 3 3 24.0 3.1 29 21 23.5 组5 6 3 3 6 0 2 4 23.7 1.0 25 23 23.0 组6 4 2 2 6 0 2 4 23.0 0.8 24 22 23.0 组7 4 2 2 6 0 3 3 22.0 2.4 25 19 22.0
XXX注射Ⅰ期临床人体耐受性试验总结报告
mean
SD 最大值 最小值 中位数 体重(kg) mean SD 最大值 最小值 中位数 既往史 无 有 过敏史 无 有
3.1.3 用药前观察指标
160.0 7.1 165 155 160.0 49.5 6.4 54 45 49.5 2 0 2 0 160.5 7.8 166 155 160.5 49.5 0.7 50 49 49.5 2 0 2 0 162.7 6.0 170 156 162.0 55.6 6.1 64 47 55.5 6 0 6 0 165.3 11.5 181 150 164.0 55.3 10.2 68 42 55.0 6 0 6 0 164.8 6.0 171 157 165.0 55.3 7.6 64 46 55.5 6 0 6 0 161.8 12.1 178 150 159.5 52.5 10.0 62 42 53.0 4 0 4 0 165.5 13.4 183 154 162.5 59.0 12.7 75 45 58.0 4 0 4 0
用药前体检、实验室检查、心电图、B超检查结果详细列表见“I期临床耐受性统计报告”,其中超过正常值范围的结果详细列于表5-7,但这些异常均无明显临床意义。
表5 用药前血液学检查异常指标
组别 受试者 指标 测定值 与正常范围比较 判别
ZHR 80.0 组1 N(%) 高 无临床意义 CCC 5.51 组2 RBC(×1012/L) 高 无临床意义 JHG 48.0 组3 N(%) 低 无临床意义 MKI 42.0 组4 L(%) 高 无临床意义
33.4 G(g/L) 高 无临床意义
PLO 47.0 组5 L(%) 高 无临床意义
NJU 6.13 RBC(×1012/L) 高 无临床意义 175 Hb(g/L) 高 无临床意义
NHU 164 组7 Hb(g/L) 高 无临床意义
BHY 76.0 N(%) 高 无临床意义
表6 用药前尿常规检查异常指标
组别 受试者 指标 结果 判别
MJU ± 组2 尿胆原 无临床意义
NHY RBC 2~4个/HP 无临床意义 WBC 1~2个/HP 无临床意义
NHU ± 组3 尿胆原 无临床意义
MKI ± 尿胆原 无临床意义 MJU ± 尿胆原 无临床意义 NHY RBC 0~1个/HP 无临床意义
NHY + 组5 尿胆原 无临床意义
NJU ± 尿胆原 无临床意义
6
XXX注射Ⅰ期临床人体耐受性试验总结报告
组6
MJU NHY WBC WBC 0~1个/HP 0~5个/HP 无临床意义 无临床意义
表7 用药前心电图、B超检查异常指标 组别 受试者 指标 组1 组7
3.1.4 用药后异常值的临床意义判断
NHT NJU NHY VGT
心电图 B超 心电图 心电图
结果 窦性心律不齐 肝右叶钙化灶 窦性心动过缓 窦性心动过缓
判断 无临床意义 无临床意义 无临床意义 无临床意义
用药前体检、实验室检查、心电图等检查结果详细列表见“I期临床耐受性统计报告”,表8-14详细列出异常检查结果,并判断其临床意义。
表8 用药前基线值正常、用药后异常的血液学指标及其临床意义判断
组别 受试者 指标 用药前值 用药后值
MMM 99 组3 PLT(×109/L)
POI 5.8 T-BIL(?mol/L) 10 D-BIL(?mol/L

) MKI 82 PLT(×109/L) 41 ALT(IU/L) LLO 79 N(%)
TRE 3.7 组4 WBC(×109/L)
BGT 80 N(%)
HHJ 41 组5 L(%)
PLO 30.7 T-BIL(?mol/L) 9.5 D-BIL(?mol/L)
POI 3.0 组6 WBC(×109/L)
OIU 31.1 T-BIL(?mol/L) POI 29.3 T-BIL(?mol/L)
PLI 3.9 组7 WBC(×109/L)
KIU 5.77 RBC(×1012/L) IIP 109 Hb(g/L) 35.8 T-BIL(?mol/L) LOI 5.72 RBC(×1012/L) 45.8 T-BIL(?mol/L) 注:用药前基线值均在正常值范围
表10 用药前正常、用药后异常的尿常规指标及其临床意义判断
判别 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 有临床意义 有临床意义 无临床意义 有临床意义 有临床意义 无临床意义 无临床意义 无临床意义 有临床意义 无临床意义 有临床意义
7
XXX注射Ⅰ期临床人体耐受性试验总结报告
组3 组4 组5 组6 组7 TRE OIU RTY UUY KJU OLI RRE NJY YTR KIU POL MKI LOI MKI MJY ERD PLI GLU 尿胆原 尿胆原 尿胆原 WBC GLU 尿胆原 RBC GLU 尿胆原 WBC 尿胆原 WBC WBC 尿胆原 尿胆原 WBC WBC 尿胆原 WBC 尿胆原 RBC WBC GLU 尿胆原 GLU GLU 尿胆原 尿胆原 - - - - - - - - - - - - - - - - - - - - - - - - - - - - - ± ± +++ +
0~1个/HP
± +
0~1个/HP
± +
0~1个/HP
+
0~1个/HP 0~1个/HP
+ ±
0~2个/HP 0~1个/HP
+++ 0~2个/HP
±
0~1个/HP 0~1个/HP
± ± ± ± +++ ± 无临床意义
无临床意义 有临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 有临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 有临床意义 无临床意义
注:用药前基线值均为正常
表13 用药前后心电图检查异常变化 组别 受试者 用药前
NHU 组1 窦性心律不齐
电轴右偏,STⅢ、avR
MKO 组3
下移<0.05mv,顺钟向转位
MMK 窦性心律不齐,窦缓 组3
BGF 组4 窦性心律不齐 NJU 组7 窦性心动过缓,心律不齐 LKJ 组7 窦性心动过缓,心律不齐 NHG 组5 正常 NVC 组7 正常
用药后
窦性心律不齐 电轴右偏,顺钟向转位 窦性心动过缓
窦性心律不齐
窦性心动过缓,心律不齐 窦性心动过缓 窦性心动过缓 窦性心动过缓
判断 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义
3.1.5 用药后实验检查异常值、不良事件与药物的相关性分析
8
XXX注射Ⅰ期临床人体耐受性试验总结报告
1.实验室和其他检查的异常值与药物的相关性:血T-BIL和D-BIL,以及尿胆原异常与药
物肯定相关,其他异常值可能与药物无关。
2.不良事件与药物相关分析:见表23。药物可能会引起头晕、皮肤不适各1例。
表14 不良事件详细列表
组别 姓名 组1 组3 组4 组6 组7
NKL BHU MNJ LKG NKI LOP VCX PLU
不良反应 头晕、胸闷 右肺干罗音 大便稀溏(2次) 脐周腹痛 咽部干痛 皮疹 头痛 头晕
程度 给药开始时间 轻 轻 轻 轻 轻 轻 轻 轻 中
2003-7-17 9:20 2003-8-4 11:00 2003-8-4 11:00 2003-8-4 10:50 2003-8-11 9:55 2003-8-11 9:55 2003-8-13 9:54
出现时间
消失时间
处理
与药物关系
2003-7-17 11:20 2003-7-17 12:20 继续用药 可能无关
2003-7-31 2003-8-4 2003-8-4 18:00 2003-8-5 9:00
继续用药 可能无关 继续用药 无法判定 继续用药 无法判定 继续用药 可能无关 继续用药 肯定有关
2003-8-4 2003-8-4 15:00 2003-8-4 13:00
2003-7-30 10:35 2003-7-31
2003-8-11 10:55 2003-8-11 13:00 2003-8-13 7:00
2003-8-11 11:50 2003-8-11 18:00 继续用药 无法判定 2003-8-11 12:30 2003-8-11 16:00 继续用药 可能有关 2003-8-13
2003-8-13
继续用药 可能无关
XSW 窦性心动过缓
3.1.6 用药后不良反应发生比例
n 组别 剂量/ml
5 2 组1
10 2 组2
20 6 组3
30 6 组4
40 6 组5
50 6 组6
60 6 组7
3.2多次给药(累积)耐受性试验 3.2.1 受试者分配
受试者根据体重和性别分配至2个剂量组,每组人数及给药剂量见表15。试验期间无退出和剔除者。
表15 受试者分组
用药剂量/ml 参加人数 退出人数 剔除人数
3.2.2 受试者一般资料
各组性别相当,年龄、身高、体重等人口统计学资料相近,无重要的既往病史和药物过敏史(见表16)。
表16 各组受试者一般资料 n
发生不良反应例数
0 0 2 1 2 2 3 发生比例 0/2 0/2 2/6 1/6 2/6 2/6 3/6
甲组 20 6 0 0 乙组 10 6 0 0
9
甲组 6 乙组 6
XXX注射Ⅰ期临床人体耐受性试验总结报告
性别
民族 职业
年龄(岁)
身高(cm)
体重(kg)
既往史
过敏史
3.2.3 用药前观察指标
男 女 汉族 非汉族 体力 非体力 mean SD 最大值 最小值 中位数 mean SD 最大值 最小值 中位数 mean SD 最大值 最小值 中位数 无 有 无 有 3 3 6 0 6 0 22.3 1.4 24 20 22.5 162.7 5.4 170 156 161.5 56.8 5.2 63 48 58.0 6 0 6 0 3 3 6 0 6 0 28.5 4.6 34 23 29.5 161.8 6.8 173 156 159.5 52.4 7.8 66 43 51.5 6 0 6 0
用药前体检、实验室检查、心电图、B超检查结果详细列表见“I期临床耐受性统计报告”,未见有临床意义的异常值。
3.2.4 用药后异常观察指标的临床意义判断
用药后体检、实验室、心电图检查等详细列表见“I期临床耐受性统计报告”,所有异常发现和不良事件见表17-23。
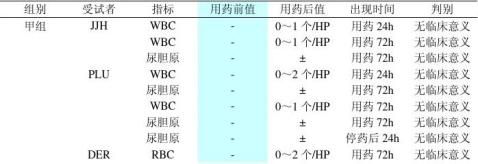
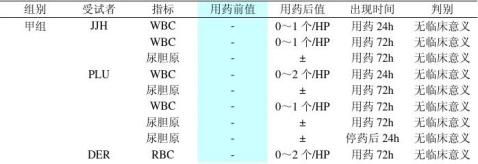
10
XXX注射Ⅰ期临床人体耐受性试验总结报告
43 甲组 CFT L(%)
7.7 D-BIL(?mol/L) MKI 1.89 RBC(×1012/L) 82 PLT(×109/L) 42 L(%) LOP 6.38 RBC(×1012/L) 162 Hb(g/L) 429 PLT(×109/L) 5.99 RBC(×1012/L) 177 Hb(g/L) 46 L(%) 41 ALT(IU/L) KJH 3.45 RBC(×1012/L) 109 Hb(g/L) 69 PLT(×109/L) OIY 6.93 RBC(×1012/L) 175 Hb(g/L) 38.0 T-BIL(?mol/L) 5.52 RBC(×1012/L) 168 Hb(g/L) 40.7 T-BIL(?mol/L) 8.5 D-BIL(?mol/L) 161 Hb(g/L) 3.64 空腹血糖(mmol/L) 42.7 T-BIL(?mol/L) 15.1 D-BIL(?mol/L)
162 乙组 BHY Hb(g/L)
7.5 D-BIL(?mol/L) HGF 91 PLT(×109/L) NKI 45 L(%) OIY 3.83 空腹血糖(mmol/L) 注:用药前基线值均在正常值范围
11
停药后24h
停药后24h 用药后24h 用药后24h 停药后24h 用药后24h 用药后24h 用药后24h 用药72h后 用药72h
后 停药后24h 停药后24h 停药后24h 停药后24h 停药后24h 用药后24h 用药后24h 用药后24h 用药后72h 用药后72h 用药后72h 用药72h后 停药后24h 停药后24h 停药后24h 停药后24h 停药后24h 停药后24h 停药后24h 停药后24h 停药后24h 无临床意义 有临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 有临床意义 无临床意义 无临床意义 有临床意义 有临床意义 无临床意义 无临床意义 有临床意义 有临床意义 无临床意义 有临床意义 无临床意义 无临床意义 无临床意义
XXX注射Ⅰ期临床人体耐受性试验总结报告
- +++ 尿胆原 用药72h - +++ 大便OB 停药后24 BJI RBC - 0~1个/HP 用药24h - ± 尿胆原 用药72h EWS WBC - 0~1个/HP 用药72h MLN - +++ 尿胆原 用药72h WBC - 0~1个/HP 停药后24h - ± 尿胆原 用药72h MJH - ± 乙组 尿胆原 用药24h
- ± 尿胆原 用药72h - ± 尿胆原 停药后24h - ++ 大便OB 用药72h DSA - +++ 尿胆原 用药24h - ± 尿胆原 用药72h MLB PRO - ± 用药24h - ± 尿胆原 用药24h - ± 尿胆原 用药72h BVC - ± 尿胆原 用药24h - ± 尿胆原 用药72h PRO - ± 停药后24h WBC - 0~2个/HP 用药72h - ± 尿胆原 用药72h - ± 大便OB 用药72h NBC WBC - 0~2个/HP 用药24h - ± 尿胆原 用药72h WBC - ++ 用药72h - 脓球 少许 用药72h - ± 尿胆原 用药72h - ± 尿胆原 停药后24 PLI PRO - ± 用药24h WBC - 3~5个/HP 用药72h - ± 尿胆原 用药72h - ± 尿胆原 用药72h 注:用药前基线值均正常
表21 用药前正常和用药后心电图检查异常的临床意义判断
组别 受试者 用药前 用药后 出现时间
PUY 甲组 正常 窦性心律不齐 停药后24h ITY 乙组 正常 窦性心动过缓 用药24h PLF 乙组 正常 窦性心动过缓 用药72h
PLF 正常 窦性心动过缓 停药后24h
有临床意义
无临床意义 无临床意义 无临床意义 无临床意义 有临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 有临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义 无临床意义
判别 无临床意义 无临床意义 无临床意义 无临床意义
3.2.5 用药后实验检查异常值、不良事件与药物的相关性分析
实验室和其他检查中的异常值中,血T-BIL、D-BIL与药物肯定相关,其他异常值可能与药
12
XXX注射Ⅰ期临床人体耐受性试验总结报告
物无关。均在4日内恢复至正常范围。
不良事件与药物相关分析见表23。药物可能会引起口干(2例)。均在24h内消失。 不良反应发生率见表24。
表23不良事件详细列表
姓名 甲组
不良反应
腹痛、脐下 2cm处压痛
轻 轻 轻 轻 程度 轻 轻
8-18 10:00 8-18 10:00
8-18 10:00
8-18 9:00 9-15 11:00 试验开始时间
2003-8-18 14:30 2003-8-23 11:00 2003-8-23 2003-8-20 5:00 2003-8-18 13:00 2003-8-18 12:00 2003-8-19 2003-8-21
(D-BIL大于正常值)
2003-9-1
继续用药
可能有关
出现时间
消失时间
8-18 14:30 2003-8-26 2003-8-26 8-20 10:00 2003-8-22 2003-8-23
处理 继续用药 继续用药 继续用药 继续用药 继续用药 继续用药
与药物关系
可能无关 可能无关 肯定无关 可能无关 可能有关 可能有关
YYT 心悸(数秒) OIY
OB+++、RBC1-4 LLK 右上腹隐痛
口干 OIY 口干
T-BIL增高、
D-BIL增高 (在正常范围内)
3.2.6 用药后不良反应发生比例 见下表。
表24 不良反应发生比例
组别 剂量/ml
20 甲组
10 乙组
n 6 6
发生不良反应例数
3 1
发生比例 3/6 1/6
结 论
1. 推荐Ⅱ期临床研究的剂量和理由
每次按10 ml/次给药,有可能引血胆红素轻度增高或尿胆原阳性,但受试者无明显不适,因此推荐II期临床试验剂量为10 ml/次;15 ml/次有可能引起血胆红素轻度增高或尿胆原阳性,风险不会太大,可根据获益/风险分析,在严密监护下尝试。20 ml/次虽然引起的血胆红素轻度增高或尿胆原阳性尚属轻度,同时无不适主诉,但发生的频度较高,慎用。
2. 不良反应的剂量
10 ml/次就有可能引起轻度不良反应,主要为血胆红素轻度增高或尿胆原阳性,但受试者无自觉症状。20 ml/次其不良反应发生例数增加。
3. 不良反应分析
血胆红素轻度增高或尿胆原阳性,且两次重复结果相近,推测是药物引起的轻度溶血所致。4日内全部恢复至正常。其他主要不良反应为口干(2例)、头晕(1例)、皮疹(1例),未经处理均在24h内消失。
13
XXX注射Ⅰ期临床人体耐受性试验总结报告
参考文献
1. SDA. 药品临床试验管理规范. 19xx年9月
2. SDA. 新药审批办法.19xx年4月
3. SDA. 中药新药研究的技术要求. 19xx年11月
4. SDA. 药品临床研究的若干规定. 20xx年7月
5. 郑筱萸主编. 中药新药临床研究指导原则(试行). 中国医药科技出版社20xx年第1版
6. 中华人民共和国中医药行业标准·中医病证诊断疗效标准. 国家中医药管理局 ZY/001.6
1995:126
14
-
临床试验总结报告的撰写[1]
临床试验总结报告的撰写定义:是反映药物临床研究设计、实施过程,并对试验结果作出分析、评价的总结性文件,是正确评价药物是否具有临床实…
-
临床试验总结报告的结构
1.题目封页2.试验药物与研究产品的名称3.研究药物的适应症4.主办者的名录5.研究的预期进度与试验安排6.试验开始数据(第一位受…
-
临床试验总结报告的设计与撰写
一、临床试验方案设计临床试验方案由研究者或申办者拟订,应符合GCP要求。研究者和申办者均应在已制定的临床试验方案上签名并签署日期。…
-
人体成分分析仪三院临床试验总结报告
医疗器械临床试验报告BCA-2A人体成分分析仪与DEXA测量对比研究总结报告产品名称:人体成分分析仪型号规格:BCA-2A实施者:…
-
临床试验总结报告的体例格式和内容要求
临床试验总结报告的体例和内容要求临床试验总结报告体例和内容要求1题目封面封面题页应包括如下内容试验题目试验药物研究产品的名称试验用…
-
临床试验总结报告的设计与撰写
一、临床试验方案设计临床试验方案由研究者或申办者拟订,应符合GCP要求。研究者和申办者均应在已制定的临床试验方案上签名并签署日期。…
-
临床试验总结报告的结构
1.题目封页2.试验药物与研究产品的名称3.研究药物的适应症4.主办者的名录5.研究的预期进度与试验安排6.试验开始数据(第一位受…
-
临床试验总结报告结构
临床试验总结报告的结构和体例格式临床试验总结报告的结构和体例格式第一部分临床试验总结报告的结构1题目封页2试验药物与研究产品的名称…
-
临床试验总结报告的撰写
临床试验总结报告的撰写定义是反映药物临床研究设计实施过程并对试验结果作出分析评价的总结性文件是正确评价药物是否具有临床实用价值有效…
-
临床总结报告
SFDA临床试验批准号20xxL00235儿肤康软膏治疗小儿湿疮亚急性湿疹风热证期临床试验总结报告试验负责单位盖章试验负责单位地址…
-
临床试验总结报告的撰写[1]
临床试验总结报告的撰写定义:是反映药物临床研究设计、实施过程,并对试验结果作出分析、评价的总结性文件,是正确评价药物是否具有临床实…