总结药化
苯巴比妥:巴比妥类镇静催眠药
构效关系(解释影响因素)
1.5位双取代才有活性,总碳数4-8最好
2.3位有甲基取代起效快
3.2位硫取代起效快
影响药物作用的因素:1)解离常数及油水分配系数的影响;2)体内代谢对药物的影响。
(1)解离常数及油水分配系数的影响。药物要在体液中转运,又要通过脂质的生物膜达到作用部位发挥药效,要求药物一定的油水分配系数和适宜的解离度。解离常数及油水分配系数的不同导致药物吸收速度不同、到达作用部位药量不同,影响药物作用强度快慢不同。如巴比妥酸解离常数较大,在生理条件下,99%以上呈离子型,无镇静催眠作用,苯巴比妥和己锁巴比妥分子型分别为50%和91%,能发挥镇静催眠作用,但己锁巴比妥比苯巴比妥作用快。油水分配系数过大,则有惊厥作用。如5位双取代总碳数超过8,导致化合物有惊厥作用。
(2)体内代谢对药物的影响。药物在体内代谢快,作用时间就短,反之较长。5位取代基的氧化是巴比妥类药物的主要代谢途径,当取代基为饱和直链烷烃或芳烃时不易代谢作用时间长,如苯巴比妥;为支链烷烃或不饱和烃基时易代谢作用时间短,如环己烯巴比妥。
地西泮(安定):苯二氮啅类镇静催眠药
苯妥因钠:抗癫痫药
普罗加比:前药型的拟氨基丁酸类抗癫痫药
盐酸氯丙嗪:吩噻嗪类抗精神病药
氟奋乃静:抗精神病药
氯普噻吨:噻吨类抗精神病药
舒必利:苯甲酰胺类抗精神病药
吗啡:生物碱类镇痛药,含酚羟基和胺,为两性化合物
哌替啶:哌啶类合成镇痛药
咖啡因:黄嘌呤类生物碱、中枢兴奋药:
硫酸阿托品:抗胆碱药 合成 CHO
+NH2+3O3AcOOClH3
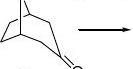
3NOAcOHO
OOOOH
麻黄碱:拟肾上腺素药 临床用途: 用于支气管哮喘、鼻塞等 苯海拉明:氨基醚类组胺H1受体拮抗剂(乘晕宁组成,优点) 马来酸氯苯那敏(扑尔敏):丙胺类组胺H1受体拮抗剂 阿斯咪唑(息斯敏):哌啶类组胺H1受体拮抗剂 普鲁卡因:局麻药 合成 O
O2
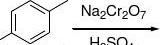
O2NN
O2N
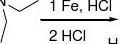
N二甲苯HCl 利多卡因(局麻药和抗心律失常)
硝苯地平:二氢吡啶类(DHP)钙拮抗剂
利舍平(利血平)作用于交感神经末梢的抗高血压药 卡托普利:血管紧张素转化酶抑制剂
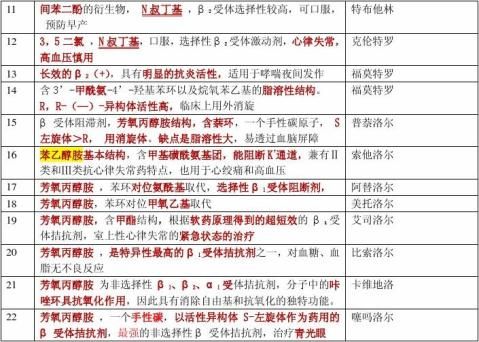
奎尼丁 普鲁卡因胺 利多卡因:抗心律失常药 普萘洛尔:β-受体阻滞剂、合成;
OOH+
OH
O
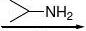
HNOH
OHN.HCl
美托洛尔:选择性β1-受体阻滞剂
氢氯噻嗪:磺胺类及苯并噻嗪类利尿药
甲苯磺丁脲:磺酰脲类口服降血糖药
第六章
抗溃疡药 雷尼替丁:呋喃类H2受体拮抗剂
奥美拉唑(洛赛克):质子泵抑制剂
昂丹司琼:止吐药
甲氧普胺、多潘立酮(吗丁啉):促动力药,止吐药
第七章
阿斯匹林:水杨酸类解热镇痛药
作用原因:
解热、镇痛、抗炎机制都与抑制前列腺素(Prostaglandine,PG)在体内的生物合成有关。研究表明前列腺素是一类致热物质,其中前列腺素E2(PGE2)的致热作用最强。前列腺素虽然本身致痛作用较弱.但能增强其它致痛物质如缓激肽、5-羟色胺、组胺等的致痛作用,加重疼痛。此外,前列腺素也是一类炎症介质。因此解热镇痛药是通过抑制花生四烯酸环氧合酶、阻断前列腺素的生物合成而达到消炎、解热、镇痛作用的。
副反应的主要原因:
(1)由于它是环氧化酶不可逆抑制剂,抑制了胃粘膜内前列腺素PGI2的生物合成,从而造成胃溃疡甚至胃出血。
(2)阿司匹林及水杨酸酸性较强,易造成对胃肠道刺激副作用。
保存及杂质来源:
1、在阿司匹林的合成过程中可能有乙酰水杨酸酐副产物生成,该微量杂质可引起过敏反应,故应检查其含量。
2、在干燥空气中较稳完,遇湿气即缓缓水解生成水杨酸和乙酸,遇碱和加热水解更快。故本品应置于密闭容器中于干燥处贮存。阿司匹林成品中由于原料残存或贮存时保管不当,可能含有过多水杨酸杂质,该杂质不仅对人体有毒性,而且较易氧化成一系列醌式有色物质,因此颜色逐渐变为淡黄、红棕甚至黑色。该杂质可采用与铁盐产生紫堇色检查。
贝诺酯(Benorilate,扑炎痛,苯乐来)是采用前药原理和拼合原理将阿司匹林和对乙酰氨基酚成酯而得对胃肠道刺激性较小,用于风湿性关节炎及其它发热所引起的疼痛,特别适合于儿童。
拼合原理指将具有协同作用的药物,经互相联结而成新药
对乙酰氨基酚:乙酰苯胺类
吲哚美辛(消炎痛):吲哚乙酸类非甾体抗炎药
第八章
环磷酰胺:前药型氮芥类抗癌药
5-氟尿嘧啶:是生物电子等排原理和代谢拮抗原理设计的抗代谢药物抗肿瘤药
紫杉醇:天然产物中发现的抗肿瘤药物
顺铂:金属配合物抗肿瘤药物
第九章
β-内酰胺抗生素:是通过抑制细菌细胞壁的合成导致细菌细胞破裂死亡,主要包括青霉素类、头孢类和非经典β-内酰胺抗生素
青霉素在长期临床应用中,充分暴露出许多缺点:对酸不稳定,只能注射给药,不能口服;抗菌谱比较狭窄,对革兰氏阳性菌效果比对革兰氏阴性菌的效果好,在使用过程中.细菌逐渐产生一些分解酶使细菌产生耐药性;有严重的过敏性反应。为了克服青霉素的诸多缺点,人们对青霉素进行结构修饰,合成大量半合成青霉素衍生物,得到一些临床效果较好的可口服、广谱、耐酶的半合成青霉素。
苯唑西林:耐酸耐酶的半合成青霉素
氨苄西林:广谱的半合成青霉素
头孢菌素与青霉素相比优点:
头孢氨苄
大环内酯类:红霉素
氨基糖苷类:链霉素:抗结核,副作用
四环素:副作用
氯霉素:副作用
第十章
磺胺嘧啶:磺胺类抗菌药物,
构效关系:
1、对氨基苯磺酰胺基是必需的基本结构,即苯环上的氨基与磺酰胺基必须处于对位,而邻位或间位异构体无抑菌活性。
2、芳伯氨基上的取代基对抑菌活性有较大的影响,多数磺胺药没有取代基,如有取代基者,必须在体内易被酶分解或还原为游离的氨基才有效,如 RCONH-、-RN=N-、-NO2等,否则无效。
3、磺酰胺基单取代可使抑菌作用增强;以杂环取代时抑菌作用明显增强,而双取代化合物一般丧失活性。
4、苯环被其它芳环取代或在苯环上引入其它基团.抑菌活性降低或丧失。
代谢拮抗原理:就是设计与生物体内基本代谢物的结构有某种程度相似的化合物,使与基本代谢物竟争性或干扰基本代谢物的被利用,或掺入生物大分子的合成之中形成伪生物大分子,导致致死合成,从而影响细胞的生长。
甲氧苄啶:抑制二氢叶酸还原酶的广谱抗菌药,常用作抗菌增效剂。
其作用机制为可逆性抑制二氢叶酸还原酶,使二氢叶酸还原为四氢叶酸的过程受阻,影响辅酶F的形成,从而影响微生物DNA、RNA及蛋白质的合成,使其生长繁殖受到抑制。与磺胺类药物联用,使细菌代谢受到双重阻断,从而使其抗菌作用增强数倍至数十倍,同时,使对细菌的耐药性减少。
诺氟沙星:喹诺酮类抗菌药
构效关系
(1)N-l位若为脂肪烃基取代时,在甲基、乙基、乙烯基、氟乙基、正丙基、羟乙基中,以乙基或与乙基体积相似的乙烯基、氟乙基抗菌活性最好。若为脂环烃取代时,在环丙基、环丁基、环戊基、环己基、l(或2)-甲基环丙基中,其抗菌作用最好的取代基为环丙基、而且其抗菌活性大于乙基衍生物。若为苯取代时,其抗菌活性与乙基相似。
(2)4-羰基和3-羧基是必需的基本结构,被其他取代基取代时活性消失。
(3)6位取代基对活性影响很大,氟最好。
(4)7位引入取代基明显增大活性,哌嗪基>二甲氨基>甲基>卤素>氢,哌嗪最好
(5)8位引入取代基对紫外光稳定,减小光毒性,以氟为最佳
利福平:抗结核药
异烟肼:抗结核药
硝酸益康唑:抗真菌药,合成
ClClClCHCOCl AlCl3ClO
ClHOClClN
N
ClCl
HO
N
ClClClClOClN
三氮唑核苷:核苷类抗病毒药
奎宁:抗疟疾药(优奎宁:碳酸乙酯奎宁,前药)
青蒿素:我国科学家发现的抗疟疾药
第十一章
甾体骨架:雌甾烷、雄甾烷、孕甾烷
己烯雌酚:雌激素
丙酸睾酮:雄激素
苯丙酸诺龙:同化激素
肾上腺皮质激素:氢化可的松、地塞米松
第十二章
VC具有烯二醇结构,显酸性。有四个光学异构体。四个异构体中以L(+)-抗坏血酸的活性最高。 VC水溶液中加入硝酸银试液,即产生银的黑色沉淀,可用于 V C的鉴别
维生素C被空气中的氧所氧化,生成去氢抗坏血酸。去氢抗坏血酸在无氧条件下就容易发生脱水和水解反应。在酸性介质中受质子催化反应速度比在碱性介质中快,进而脱酸生成呋喃甲醛,呋喃甲醛易于聚合而呈现黄色斑点。这是本品在生产贮存过程中变色的主要原因。
第十三章
特异性结构药物是指药物的作用依赖于药物分子的特异的化学结构,及其按某种特异的空间相互关系排列。
影响药物产生药效的决定因素:
1、药物必须以一定的浓度到达作用部位,才能产生应有的药效。
2、在作用部位,药物和受体形成复合物,通过复合物的作用,产生生物化学和生物物理的变化。 举例说明理化性质对药效的影响 (见苯巴比妥)
影响药效的化学结构因素:
①药物的基本结构,②电子云密度的分布,③键合特性,④立体结构。
药物分子和受体间的结合形式有哪些
药物分子和受体间的结合通过静电相互作用、氢键,范德华力,疏水结合,电荷转移物,以及共价键形式作用。
立体结构对药效的影响:药物的手性中心在受体结合中的部位直接影响到生理活性。如药物的手性中心不在受体结合的部位,则对映体的作用完全相同;如药物的手性中心在受体结合的部位,则对映体或者作用强弱不同,或者作用方式不同,如由抑制剂变成了激动剂。
药效构象:化合物与受体间相互结合时的构象
先导化合物是通过各种途径和方法得到的具有某种生物或药理活性的化合物。
先导化合物的发现途径:
1、从天然活性物质中筛选和发现先导化合物
2、以生物化学或药理学为基础发现先导化台物
3、以现有的药物作为新药研究的基础
4、用普筛方法发现先导化合物
合理药物设计(Rational Drug Design):根据对生理病理的了解来研究新药 ,通常是针对与该生理活动有关的酶和受体来设计药物。
Me-Too药物特指具有自己的知识产权的药物,其药效和同类的突破性药物相当。
先导化合物的优化方法:生物电子等排原理、前药原理、软药原理和定量构效关系研究
生物电子等排体:具有相似的物理及化学性质,又能产生大致相似或相关的或相反的生物活性的基团或取代基。
前药(Prodrug):是指一些无药理活性的化合物,但是这些化合物在生物体内可经过代谢的生物转化或化学的途径,被转化为活性的药物。
前药修饰的主要用途:
(1)增加药物的代谢稳定性,延长药物的作用时间
(2)改善药物的吸收和分布,提高作用选择性
〔3)增加药物的溶解度,增加药物的化学稳定性,适应剂型需要
(4)减低毒性或不良反应及消除药物不适宜的性质,使病入容易接受
硬药是指具有发挥药物作用所必需的结构特征的化合物,但该化合物不发生代谢或化学转化,可避免产生不必要的毒性代谢产物可以增加药物的活性。
软药是本身具有治疗作用的药物,在体内作用后,经预料的和可控制的代谢作用,转变成无活性和无毒性的化合物。
定量构效关系(QSAR)是一种新药设计研究方法,是用一定的数学模型对分子的化学结构与其生物效应间的关系进行定量解析,从而寻找出结构与活性间的量变规律。
第二篇:药化总结表格9-15章

-
药物化学总结
第一章麻醉药分类:苯甲酸酯类、酰基苯胺类等局麻药的构效关系(1)亲脂性部分1苯环的邻对位引入给电子取代基,如氨基、羟基、烷氧基时,…
-
药物化学总结
?抗过敏药分类及代表药物一.组胺H1受体拮抗剂和抗过敏药1.经典H1受体拮抗剂:乙二胺类:芬苯扎胺,曲吡那敏。氨基醚类:苯海拉明,…
-
药化总结
课件中的思考题第十四章心脏疾病药物和血脂调节药1抗心律失常药可分为哪几大类各举一例答I类钠通道阻滞剂IA类奎尼丁IB类美西律利多卡…
-
药物化学总结
药物化学总结第一章麻醉药第一节全身麻醉药1吸入麻醉药氟烷2溴2氯111三氟乙烷起效苏醒快作用弱全麻及诱导麻醉性质1氧瓶燃烧2加入硫…
-
药物化学知识点总结
药物化学知识点总结不含结构式师兄只能帮你们到这里了需要的自己粘贴来源田牧的日志前言这是本人总结的药物化学知识点请仔细核对一下是否正…
-
药物化学总结
第一章麻醉药分类:苯甲酸酯类、酰基苯胺类等局麻药的构效关系(1)亲脂性部分1苯环的邻对位引入给电子取代基,如氨基、羟基、烷氧基时,…