��ѧ�л���ѧ��ĩ��ϰ�ܽ�
�л���ѧ��ĩ��ϰ�ܽ�
���γ̵�ѧϰ�����������ֽ�ȫ����ص����ݰ��������ṹ���ۡ�������Ӧ��������ת�����ϳɷ���������ȼ���ר������ܽ���ɣ���ͬѧ�Ǹ�ϰʱ�ο���
һ���л������������
������ѧϰ�л���ѧ�ġ����ԡ�����ˣ�Ҫ��ѧϰ�߱������ա��л��������������������ϰ��������ϵͳ�����ȷ�����Ҫ���ܶԳ����л�������д����ȷ�����ƻ��������д���ṹʽ����ʽ��
1�� ��������д Ҫ������һЩ���������������Ļ�����Ľṹʽ���磺ľ�����ʴ���
���͡�ʯ̿�ᡢ���ᡢˮ��ȩ��ˮ���ᡢ�ȷ¡����ᡢ��ζ�ᡢ����ᡢ�������ʰ��ᡢ�����ᡢ�Ȱ��ᡢ�Ͷ�ȩ�������ǡ����ǵȡ���Ӧ��ϤһЩ��������д����Ʒ�����������Ļ�����磺RNA��DNA����˾ƥ�֡�ú���������ն������������֡�����Ϣʹ����Ŷ��ȡ�
2��ϰ�������� Ҫ�����ա������졢�¡����������١��塢��������ͷ�ĺ��弰�÷���
���ճ��������Ľṹ���磺ϩ��������ϩ��������������������춡�����嶡�����л��ȡ�
3��ϵͳ������ ϵͳ���������л��������������ص㣬�����������ո�����������ԭ����������������ǻ����������칹�塢��ѧ�칹��Ͷ�����Ż�������������ѵ㣬Ӧ�������ӡ�Ҫ�μ�����������ѭ�ġ��������
��1�� �������칹������� ϩ�������칹�����������˳������Z��E���ַ�����
�Ļ����������˳����ʾ��Ҳ������Z��E��ʾ����˳����ʾʱ����ͬ��ԭ�ӻ������˫��̼ԭ��ͬ���Ϊ˳ʽ����֮Ϊ��ʽ�����˫��̼ԭ���������ĸ����Ŷ�����ͬʱ��������˳����ʾ��ֻ����Z��E��ʾ�����ա�������Ƚ����Ի��ŵ�����˳���Ż�����˫��̼ԭ��ͬ���ΪZ�ͣ���֮ΪE�͡�����ע�⣬˳������Z��E�����ֲ�ͬ�ı�ʾ�����������ڱ�Ȼ��������ϵ���еĻ����������˳����ʾ��Ҳ������Z��E��ʾ��˳ʽ�IJ�һ����Z�ͣ���ʽ�IJ�һ����E�͡����磺
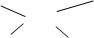 CH3-CH2 Br
CH3-CH2 Br
C=C ����ʽ��Z�ͣ�
H CH2-CH3
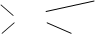 CH3-CH2 CH3
CH3-CH2 CH3
C=C (��ʽ��E��)
H CH2-CH3
֬��������Ҳ����˳���칹�壬����ȡ�����ڻ�ƽ���ͬ��Ϊ˳ʽ����֮Ϊ��ʽ��
��2������ѧ�칹������� ��ѧ�칹��Ĺ��������ֱ�ʾ����D��L��R��S��
D ��L��Ƿ��Ը���ȩΪ������һ���ľ����ԣ���Щ���������ȷ���������ȩ�ṹ�Ķ�Ӧ��ϵ����ˣ��������Ӧ��R��S��Ƿ������Ǹ�������̼ԭ�������ĸ���ͬԭ�ӻ�����ڿռ������˳���ǵġ���ѧ�칹��һ����ͶӰʽ��ʾ��Ҫ���շ�Ъ��ͶӰʽ��ͶӰԭ���͵��жϷ��������磺
COOH ����ͶӰʽ�жϹ��ͣ�����Ҫ��ȷ��
 H NH2 ��ͶӰʽ�У���������������ǰ ��
H NH2 ��ͶӰʽ�У���������������ǰ ��
CH2-CH3 ����������������ٸ��ݡ�����
������������̼ԭ�������ĸ����ŵ�����˳������ʽ�У�
��NH2>��COOH>��CH2-CH3>��H ������С������ԭ����Ϊ��̼ԭ��Ϊ���ĵ��������嶥�ˣ�������������Ϊ��������ײ������εĽǶ�����������ײ��˷����������ţ��Ӵ�С��˳ʱ��ΪR����ʱ��ΪS ������ʽ�У��ӣ�NH2 ��COOH ��CH2-CH3Ϊ˳ʱ�뷽�����ͶӰʽ�������Ļ�����ΪR���ͣ�����ΪR-2-�������ᡣ
��3��  ��˫�����Ż���������� ˫�����źͶ�����Ż�����������ؼ���ȷ��
��˫�����Ż���������� ˫�����źͶ�����Ż�����������ؼ���ȷ��
ĸ�塣�����������¼��������
�� ��±�غ����������������Ų���ʱ����±�غ�������Ϊȡ����������������Ϊĸ�塣
�� ��˫�����ǻ����ʻ����Ȼ�����ʱ������ϩ��Ϊĸ�壬�����Դ���ȩ��ͪ������Ϊĸ�塣
�� ���ǻ����ʻ�����ʱ����ȩ��ͪΪĸ�塣
�� ���ʻ����Ȼ�����ʱ��������Ϊĸ�塣
�� ��˫������������ʱ��Ӧѡ��Ⱥ���˫���ֺ����������̼��Ϊ���������ʱ��˫���������Ծ����ܵ͵����֣����˫����������λ������ͬ����Ӧ��˫������ͱ�š�
4���ӻ������������ ���ڴ��ӻ�ĸ��������������������������ԣ�һ��������뷨��Ҫע��ȡ�����ı�š�
�����л�������Ľṹ����
1��̼ԭ�ӵ��ӻ����ۼ� Ҫ�������ӻ��ĸ��̼ԭ�ӵ������ӻ���ʽSP��SP2��SP3�Լ������ӻ���ʽ���ص㣬 ���ж��л�����������и�̼ԭ�ӵ��ӻ���ʽ�����զ� ���ͦ� �����γɼ����ǵ����𣬹���� ���ͷ����� ���������Լ�˫������������ʲô����ɵġ����ղ�ͬ�ṹ��̼�����Ӽ����ɻ����ȶ��ԡ�
2��ͬ���칹 ͬ���칹�������л������������֮һ������ʽ��ͬ�������и�ԭ�ӵ����д���ͷ�ʽ��ͬ�Ļ����ﻥΪͬ���칹�塣ͬ���칹�����Ϳɹ������£�
 ̼���칹
̼���칹
λ���칹
 �����칹 �������칹
�����칹 �������칹
�����칹
 ͬ���칹 ˳���칹
ͬ���칹 ˳���칹
 �����칹
�����칹
�����칹 �����칹����ӳ�칹��
�����칹
̼���칹������̼�Ǽܣ�̼�����IJ�ͬ�������ģ����磬������춡�顣
λ���칹������ȡ�������������̼�����ϵ�λ�ò�ͬ�������ģ����磬��������2-������
�������칹�����ڹ����ŵIJ�ͬ�������ģ����磬�����������ѡ�
�����칹�����ڲ�ͬ������֮��Ѹ�ٻ�����γɵĿ����칹���������磬
 CH3��C=CH2
CH3��C=CH2 CH3��C��CH3
CH3��C��CH3
˳���칹������˫���Ĵ���ʹ������ijЩԭ�ӻ�����ڿռ�λ�õIJ�ͬ�������ģ����磬˳-2-��ϩ�ͷ�-2-��ϩ��
��ӳ�칹�����ڷ����к�������̼ԭ�ӣ������ĸ���ͬԭ�ӻ���ţ���ԭ���ڿռ�����з�ʽ��ͬ��ʹ��������֮�����ʵ���뾵��Ĺ�ϵ����Ϊ��ӳ�칹�����磺

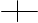 COOH COOH
COOH COOH
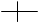 H OH HO H
H OH HO H
CH3 CH3
�����칹������Χ�ƦҼ���ת�������ķ�����ԭ�ӻ�����ڿռ䲻ͬ�����з�ʽ�й����칹�����磺1-������ȫ�ص�����Ͷ�λ���湹��

3.����ЧӦ ����ЧӦ�����յ�ЧӦ����ЧӦ�����ࡣ
�յ�ЧӦ�����ڷ�����ԭ�ӵĵ縺�Բ�ͬ��������һ�ּ���ЧӦ���ئҼ��ɽ���Զ���ݣ�����ԽԶӰ��ԽС���������������յ�ЧӦ�������յ�ЧӦ��
����ЧӦ���ڹ�����ϵ�����ڵ��ӵ������������ЧӦ���ع�����ϵ�Լ��Խ���ķ�ʽ���ݣ�����̼���������������������������ӹ���ЧӦ�����ӹ���ЧӦ������ЧӦ����ʽ�Ц�-�й���繲��ϩ������������p-�й����ϩ����̼�����ӣ���
Ҫ�������յ�ЧӦ����ЧӦ������ԭ���ص㣬���õ���ЧӦ����̼�����ӡ�̼�����Ӻ���������ȶ��ԣ�ȡ���������ᡢ�����Ե�Ӱ�켰�����Ե�Ӱ�죻���Ͳ��Գ�ϩ��������ӳ�ʱ����ѭ�����Ϲ���±�����ʹ���������ʱ����ѭ�IJ����ɷ����ͬ�ṹ��±����������ȡ����Ӧ�Ļ��ԡ��ʻ�����������˼ӳɷ�Ӧ�Ļ��Եȡ�
�����л�������Ļ�����Ӧ
1���ӳɷ�Ӧ�����ݷ�Ӧ���̲�ͬ��Ϊ��ӳɡ��˼ӳɺ�������ӳɡ�
��1�� ����ӳɣ������Լ��Ľ��������еļӳɷ�Ӧ��Ҫ�����ղ��Գ�ϩ������
��ӳɷ�Ӧʱ����ѭ�����Ϲ����Լ��д�����˵IJ��ּӵ�����϶��˫��̼ԭ���ϣ������Բ��ּӵ�������ٵ�˫��̼ԭ���ϡ�ϩ����±�ء�±���⡢���ᡢ��±�ᡢˮ��Ȳ����±�ء�±���⡢ˮ�Լ�����˫ϩ��1��2��1��4�ӳɶ�����ӳɷ�Ӧ��ϩ��������ӳɷ�Ӧʱ��˫���ϵ������ܶ�Խ��ӦԽ�����С�
��2�����˼ӳɣ������Լ����������еļӳɷ�Ӧ��Ҫ�������Լ��ĸ���˼ӳɷ�Ӧ�����̣��ӳɼ��ӳɩ�����������ͬ�ṹ���ʻ�����������˼ӳɷ�Ӧ�Ļ���˳��Ӱ�췴Ӧ���Ե����ء��ʻ��������������ᡢ���������ơ����������Լ���������������ļӳɶ����˼ӳɷ�Ӧ�� �ʻ�����������˼ӳɷ�Ӧ�Ļ���˳��Ϊ��
HCHO>CH3CHO>RCHO>C6H5CHO>CH3COCH3>RCOCH3>C6H5COCH3>C6H5COC6H5
��3�������ɻ��ӳɣ������ɻ����������еļӳɷ�Ӧ��ϩ���ڹ���������������廯����еļӳ������ɻ��ӳɡ����Գ�ϩ�����廯��������ɻ��ӳ�ʱ�õ������Ϲ���IJ������ӵ�������ٵ�˫��̼ԭ���ϡ�
�ӳɷ�Ӧ��������������֮�⣬���в��������Ĵ��⻯�������ϩ��˫ϩ�ϳɵȡ�
2��������Ӧ ��һ���������������ȥ��С���ӣ���H2O��HX��NH3�� ���γ� ˫���������ķ�Ӧ��������Ӧ��±������±����ʹ���ˮ����Ҫ��������Ӧ��
��1����±������±���⣺±������������Ӧ����ǿ���������½��С���ͬ�ṹ��±��������������Ӧ�Ļ���˳��Ϊ������>����>һ����Ҫ����±��������������Ӧʱ����ѭ�IJ����ɷ����±�����в�ֻ����һ����̼ʱ������ʱ��ȥ�����ٵ���̼�ϵ���ԭ�ӡ�Ҫע�⣬±������������ˮ���Ǿ�����Ӧ��
��2������������������������Ӧ��ǿ���������½��У���������Ҳ��ѭ�����ɷ����Ҫ���ղ�ͬ�ṹ�Ĵ�����������Ӧ�Ļ���˳���崼>�ٴ�>������
3��ȡ����Ӧ ���ݷ�Ӧ���̵IJ�ͬ�ɷ�Ϊ��ȡ������ȡ���������ȡ����
�š���ȡ�����������Լ��Ľ�����������ȡ����Ӧ����ȡ����Ӧ�������ϵ�±�����������ǻ���������������������Լ��ص��ε�ż�Ϸ�Ӧ�ȣ�������ȡ����Ӧ�������ͷ����ӻ���Ҳ�ܷ�����ȡ����Ӧ��Ҫע�ⱽ�������¶ۻ���ʱ���ܽ��и��Ϸ�Ӧ�������Ͻ��������ʱ�ᷢ���칹���������������Ͻ�����ȡ����Ӧ�Ĺ��ɣ���һ��ȡ������Ҫ�����λ���ڶ���ȡ�����ǽ���ͬ�������컷��ԭȡ�����Ķ�λЧӦ������������Ա����Ա�����ӻ����������ȡ����Ӧ�Լ������뱽���ȽϽ�����ȡ����Ӧ���ԵIJ��죬ૡ���ԡ�����������ȡ����Ӧ�Ļ��Աȱ�����ऱȱ�С��
�ơ���ȡ�� �����Լ��Ľ����������ȡ����Ӧ����ȡ����Ӧ��±������ˮ�⡢���⡢��⡢���⣬������±��ķ�Ӧ���Ѽ��Ķ��ѣ������������ˮ�⡢���⡢����ȶ�����ȡ����Ӧ��±��������ȡ����Ӧ�ɰ��������̽��У����������̣�SN1����˫�������̣�SN2����һ��±�����װ�SN2���̷�Ӧ������±����һ�㰴SN1���̷�Ӧ������±�������������֮��Ҫ�����ⷴӦ���̵Ļ��������ղ�ͬ±����������ȡ����Ӧ�Ļ��ԡ�Ҫע�⣬�ڼ���������±������ȡ���������ǻ��ྺ���ķ�Ӧ������±����������������һ��±��������ȡ����ǿ�����ܼ�����ˮ��������ȡ�������������ܼ����紼����ǿ��紼�ƣ�����������������������������
�ǡ����ɻ�ȡ���������ɻ������������е�ȡ�������ɻ�ȡ����������±����ϩ����������Ħ�±�������ɻ�ȡ����Ӧ����Ӧ�����Ǹ��¡����ջ����������ڡ����ɻ����ȶ��Ժ�����̼ԭ���������������Ŀ�йأ����Խ�࣬�ȶ���Խ�����ɻ����ȶ�����Ϊ��
����>����>һ��>·CH3
4��������ԭ��Ӧ ����������Ӧ�ͻ�ԭ��Ӧ�������͡�
�š�������Ӧ ϩ��Ȳ�����������Լ������ӡ�ȩ��ͪ�ȶ�����������ӦҪ���ռ��ֳ��õ����������������ء��ظ���ص�������Һ���������������������Լ������Լ�������Լ�����±���Ƶȡ�����������Ӧ��ʵ���е�Ӧ�ã�����������������Ʋ�ϩ���Ľṹ�������Լ�������Լ�����������������ȩ��ͪ�ȡ�
�ơ���ԭ��Ӧ ���������Ĵ��⻯��ȩ��ͪ�����ἰ����ԭΪ������������ԭΪ�����ȶ��ǻ�ԭ��Ӧ��Ҫ���ռ��ֳ��õĻ�ԭ������H2/Ni�� Na+C2H5OH��Fe+HCl��NaBH4���� ��LiAlH4�������/��������ȣ�ע������������ṩ�������ӵĻ�ԭ����ֻ���ʻ�ѡ����⣬��˫�����������������á���Ҫ�����ʻ���ԭΪ�Ǽ������ַ�����ע�⣬���п�����ɭ��ԭʱ��Ӧ������в��ܴ��ڶ������еĻ��ţ��紼�ǻ���˫���ȣ����������������ԭ���������Ľ���ʱ����Ӧ������в��ܴ��жԼ����еĻ��ţ���±�صȡ�
5�����Ϸ�Ӧ ��Ҫ������ȩ���Ϻ������ϡ�
��1����ȩ���� ���Ц����ȩ��ϡ�����������������ǻ�ȩ���˻����ﲻ�ȶ�
����������ˮ������������������ȩ����ˣ��˷�Ӧ����������̼���Ʊ�������������ȩ��Ҫ��������ȩ���ϵķ�Ӧ������
��2������ɭ������ ���Ц��������ǿ�������·�������ɭ�����ϣ���������֮����ȥһ���Ӵ����ɦ�ͪ������Ҫ���շ�Ӧ��������ʵ���е�Ӧ�ã��л��ϳ��й㷺Ӧ�õ�����������������ͨ���˷�Ӧ�Ʊ��ġ�
���������������͵ķ�Ӧ֮�⣬��Ҫ�������ص�����Ӧ�������ص��ε�ȡ����Ӧ�����ȷ�Ӧ�ȣ�ע�ⷴӦ���������P����ʵ���е�Ӧ�á�
�ġ��л��������ת�����ϳɷ���
Ҫ�������л���������������֮���ת����ϵ������������̼�ķ������ڴ˻�������Ƽ��л�������ĺϳ�·�ߡ��������ձ����и������������������Ȳ�������������ȩ���ϡ������Լ����ȶ���������̼����Ȳ����������������Լ����������ص��ε����л��ϳ���Ӧ�÷dz��㷺��
1��Ȳ��������� ����Ȳ���Ȳ���백�������õ�Ȳ�ƣ�Ȳ���벮±������Ӧ�õ����ȡ����Ȳ�����˷�Ӧ������̼�����Ʊ���Ȳ����
2�������Լ��� �����Լ����л��ϳ���Ӧ�ü�Ϊ�㷺�����뻷�����顢ȩ��ͪ������Ӧ�������Ʊ���ͬ�ṹ�Ĵ��ȡ���Щ��Ӧ�ȿ�����̼�����ֿ��γ�����Ĺ����š�
3���ص���ȡ���� �����ص��ε��ص����ɱ���ԭ�ӡ�±�ء��ǻ������ȡ�������ڱ�����ԭ��ȡ������λЧӦ��Ӱ���ʹijЩ���Ų���ֱ�����뱽��ʱ���ɲ����ص���ȡ���ķ�����Ҫע�ⱻ��ͬ����ȡ��ʱ�ķ�Ӧ������
�塢�л�������ļ���
ϩ������ϩ��Ȳ����������Ԫ��֬������������Ȼ�̼��Һ������ɫ
����Ȳ���Ȳ�������������Ȼ���ͭ�İ���Һ������Ȳ������ɫ������Ȳ����ͭ��ɫ������
±�������������Ĵ���Һ������±������������ͬ�ṹ��±�������ɳ������ٶȲ�ͬ����±������ϩ��ʽ±������죬��±������֮����±��������Ȳų��ֳ�����
����������Ʒ�Ӧ�ų�����������6��̼ԭ�����µĴ�������¬��˹�Լ������١��崼���崼���̱���ǣ��ٴ����ú����ǣ��������ú�Ҳ�ޱ仯��
�ӻ�ϩ�����������Ȼ�����Һ������ɫ����������ˮ�������屽�Ӱ�ɫ������
 �ʻ������2��4-���������£�������ɫ��Ⱥ�ɫ����������ȩ��ͪ�������Լ���ȩ��������������ͪ���ܣ�������ȩ��֬��ȩ��ͪ��֬��ȩ��������Լ���֬��ȩ����ש��ɫ��������ͪ�ͷ���ȩ���ܣ������ͪ�;���CH3��CH���ṹ�Ĵ��õ������������Һ��
�ʻ������2��4-���������£�������ɫ��Ⱥ�ɫ����������ȩ��ͪ�������Լ���ȩ��������������ͪ���ܣ�������ȩ��֬��ȩ��ͪ��֬��ȩ��������Լ���֬��ȩ����ש��ɫ��������ͪ�ͷ���ȩ���ܣ������ͪ�;���CH3��CH���ṹ�Ĵ��õ������������Һ��
���ɻ�ɫ�ĵ�³�����
����������Լ���������������������������ܡ�
���������١��尷�����ٷ���
1�� �ñ������Ȼ�Լױ������ȣ���NaOH��Һ�з�Ӧ���������ɵIJ�������NaOH���ٰ����ɵIJ��ﲻ����NaOH��Һ���尷��������Ӧ��
2�� ��NaNO2+HCl��
֬�����������ų��������ٰ����ɻ�ɫ��״��尷����Ӧ��
���㰷�����������ص��Σ��ٰ����ɻ�ɫ��״��尷������ɫ���塣
�ǣ�����������ǣ�����ˮ����������ʹ��ˮ��ɫ�������Dz��ܡ�
��ѿ�������ǣ��������Լ�������Լ�����ѿ�ǿ�����������ש��ɫ�����������Dz��ܡ�
����ʵ��
Ҫ������ʵ���һ���֪ʶ���л���ѧʵ��Ļ����������ܡ��������ؽᾧ�����۵����õ�װ�ü�������ע�����Һ������ʱ���õ�������װ�á���ƿ������Һ���������¶ȼư�װ��λ�á��ŷ�ʯ��ʱ�䡢����ͬ�е�Һ�廯����ļ��ȷ�ʽ���������ܵ�ѡ��ϴ��Һ�廯�������õ�����������ʱע�������������������װ�ã�����ƿ���ܾ����ӵ�ѡ���Լ�ʵ���ҷ��������ʶ�ȡ�
����������ר��֮�⣬��Ӧ���ո��µĻ���������ʡ�����ĺ��塢���������������ʵı仯�����Լ�ҽҩ�����������о����漰�����л���ѧ������ʶ�ȡ�
�ڶ�ƪ���л���ѧ��ϰ�е�30�����������ܽ�����
�л���ѧ��ϰ�е�30�����������ܽ����� 1������Ϊ�л������ȼ�ա�
�����Ȼ�̼����ȼ�գ������Ǹ�Ч������
2������Ϊ���ȼ��������ֽṹ��
��Ϊ���鲻��ƽ��ṹ������������ṹ���ʶ��ȼ���ֻ��һ�ֽṹ��
3������Ϊ̼ԭ��������4�����ڳ��³�ѹ�¶���Һ�����塣
�����������⣬�е�9.5�棬���塣
4������Ϊ�������Ը��������Һȥ�������е���ϩ��
��ϩ�����Ը���������������������̼���ʲ��ܴﵽ����Ŀ�ģ��������ü�ʯ�Ҵ�����
5������Ϊ˫������С�����ȶ������ѡ�
��ʵ��˫����ֻ��һ������������������
6������Ϊϩ������ʹ��ˮ��ɫ��
���ϩ������ˮ�в�����ʹ����ɫ��������������Ȼ�̼��Һʱȴ��ʹ����ɫ����Ϊ����Խ��Խ��������ˮ������Ӵ���
7������Ϊ����ϩ�Ǵ����
����ϩ�ǻ�����Ϊ���ǵ���Է�������������
8������Ϊ��Ȳ����ˮ�����Ը��������Һ��Ӧ�����ʱ���ϩ�졣
������ʵ˵����Ȳʹ������ɫ���ٶȱ���ϩ���öࡣ
9������Ϊ��״̼������ˮ��Ӧ������Ȳ��������ȣ��������շ�������
���ڵ�ʯ��ˮ��Ӧ���ٶȺܿ죬�����ƣ�ͬʱ�ų��������ȣ���Ӧ�в����ĺ�״�ﻹ���ܶ�������©����ײ�����֮��Ŀ�϶���ʲ��������շ�������
10������Ϊ����������ڹ������ܷ���ȡ����Ӧ���ʱ��������ڹ���(������)������Ҳ�ܷ���ȡ���� ���������������������·������Ǽӳɷ�Ӧ���������Ȼ����顣
11������Ϊ������ˮ����Ӧ��������Ϻ�����������
��Ȼ���߲���Ӧ����������ȡˮ�е��壬�ʿ���ˮ����ɫ��dz����ȥ���������Ϊ�Ⱥ�ɫ��
12������Ϊ�����Ը��������Һ���Գ�ȥ���еļױ���
�ױ��������ɱ����ᣬ�������������ڱ������ѷ��롣Ӧ��������������Һʹ������ת��Ϊ������ˮ�ı������ƣ�Ȼ���Һ��
13������Ϊʯ�ͷ����õ������Ϊ�����
���������һ���е㷶Χ�ڵ���֣���Ϊ����
14������Ϊ�����Ը��������Һ������ֱ�����ͺ��ѻ����͡�
ֱ�������к��н϶�ı���ͬϵ��;���߲��������Ը�����ؼ���
15������Ϊ±����һ���ܷ�����ȥ��Ӧ��
16������Ϊ�������ǻ��������л���һ���Ǵ��ࡣ
�����Ƿ��ࡣ
17������Ϊ�����ǹ��壬��������ˮ���ܽ�Ȳ��ʴ������Ӵ�ˮ������ʱ�������������ù��˵ķ������롣
������ˮ���г�����������������������ˮ������ʱ�������ֲַ������²��DZ���������������ˮ����Һ���ϲ��෴����Ӧ�÷�Һ�ķ������뱽�ӡ�
18������Ϊ�Ҵ���Һ�壬�������ǹ��壬���Ӳ�������Ʒ�Ӧ��
���屽���䲻���Ʒ�Ӧ�����������ۻ����������Ʒ�Ӧ���ұ��Ҵ����Ʒ�Ӧ�����ҡ�
19������Ϊ���ӵ����Ա�̼������̼��ֻ��ʹ��ɫʯ����Һ��죬���Ƕ϶�����һ������ʹָʾ����ɫ��
������ǿ�����١���ȴ�С�������ͱ�����Һ�ȱ���̼���Ũ�ȴ�Ũ�Ƚϴ�ı�����Һ��ʹʯ����Һ��졣
20������Ϊ�������Ա�̼�������ʱ��Ӳ�����̼������Һ��Ӧ��
���ӵĵ���̶����̼��С����ȴ��̼��������Ӵ������ɸ��ֽ���ɿ�֪�����Ӻ�̼������Һ�ܷ�Ӧ���ɱ����ƺ�̼������
��
21������Ϊ����ȥ���еı��ӿ������м�������Ũ��ˮ���ٰ����ɵij������˳�ȥ��
��������ˮ��Ӧ��������ױ���ȡ�����У��������ɵ����屽���䲻����ˮ��ȴ�����ڱ������Բ��ܴﵽĿ�ġ�
22������Ϊ��������ˮ��Ӧ�������屽�ӣ��ױ�����������TNT�����ƶϹ�ҵ��ȡ��ζ��(����������)��ͨ�����ӵ�ֱ�������Ƶõġ�
���ƶϺ����˱����ױ����������ʡ������м���Ũ����ʱ���ֱ��ӱ��������������ʼ��͡���ҵ��һ�����ɶ������ȱ�����������ˮ���Ƶÿ�ζ�ᡣ
23������Ϊֻ�д����γ��������Ӳ����γ�����
����Ҳ���γɶ�Ӧ�������簢˾ƥ�־��Ƿ�����������ڴ����ԣ��ӳ��������ѣ�ͨ�����������������ȷ�Ӧ��������
24������Ϊ��һ���ɷ���ȥ��������
��̼Ϊ���Ĵ����ܷ���ȥ�������������촼��
25������Ϊ����һԪ��������һ������ȩ��
���ǻ�����̼����ʱ��������ͪ����2-������
26������Ϊ��һ���ܷ�����ȥ��Ӧ��
�״�����̼����Ĵ����ܷ�����ȥ��Ӧ��
27������Ϊ���봼��Ӧ���ɵ��л���һ��������
�Ҵ��������ᷴӦ���ɵ�����������±��������������
28������Ϊ������Ӧһ�����ǡ���ȥ�ǻ���ȥ�⡱��
�Ҵ�����������ᷴӦ��һ���Ǵ�ȥ�ǻ���ȥ�⡣
29������Ϊ���Ƿ����к����Ȼ����л���һ�������ᣬ����ʹʯ���졣
Ӳ֬���ʹʯ���졣
30������Ϊ��ʹ�л�����������������ķ�Ӧһ����������Ӧ��
�Ҵ���Ũ���ᷢ��������Ӧ����������������
- ��ѧ�л���ѧ֪ʶ���ܽ������
- ��ѧ�л���ѧ��ĩ��ϰ�ܽ�
- ��ѧ�л���ѧ�����ܽ�
- �л���ѧ�Ƶ��ܽ�
- ��ѧ�л���ѧ��ĩ��ϰ�ܽ�
- �л���ѧ��������ʽ�ܽ�
-
�л���ѧ����ʽ�ܽ�
�����л���ѧ����ʽ�ܽ�һ����1.��������ͨʽ��CnH2n-2��1��������Ӧ�����ȼ�գ�CH��ȼ4+2O2���鲻��ʹ���Ը��������Һ��
-
�л���ѧ��������ʽ�ܽ�
һ��֬����1.������ѧ���ʣ�����ǿ�ᡢǿ�ǿ��������ǿ��ԭ����Ӧ������ʹ������Ȼ�̼��Һ�����Ը��������ˮ��HC��CH+H2O����
- ��ѧ�л���ѧ��ĩ��ϰ�ܽ�
-
��ѧ �л���ѧ ��ϰ-�ܽ�
�л���ѧ��ϰ�ܽ��л�������Ļ�����Ӧ�ӳɷ�Ӧ�����ݷ�Ӧ���̲�ͬ��Ϊ��ӳɡ��˼ӳɺ�������ӳɡ���ӳɣ������Լ��Ľ��������С�
- ��ѧ�л���ѧ֪ʶ���ܽ������