白细胞计数实验报告
白细胞计数和分类
目的:掌握血涂片制备的操作要领、瑞氏染色方法、正常外周血五种白细胞形态
特点、
原理:各种白细胞必须经过染色,才易于区分其类别。常用者为瑞氏(Wright’s)
染色法和姬姆萨(Giemsa)染色法。测定外周血液中各种白细胞的相对比值,以观察其数量、形态和质量变化,通过显微镜观察染色血涂片,计算血液中各类白细胞的百分率,称为白细胞分类计数。利用白细胞总数和各类白细胞的百分率,即可计算每mm3血液中各类白细胞的绝对值。
实验器材: 香柏油、盖玻片、推片、采血针或注射器、小滴管、消毒棉球、瑞
氏染液、pH6.4~6.8磷酸盐缓冲液、蒸馏水、计数板及专用盖片、针 1ml刻度移液管、1ml EP管、光学显微镜。20ul刻度移液管、推片
实验步骤
白细胞分类
1.血片制作:
取一滴血,滴于洁净无油脂的玻片一端。左手持玻片,右手再取边缘光滑的另一玻片作为推片。将推片边缘置于血滴前方,然后向后拉,当与血滴接触后,血即均匀附在二玻片之间。此后以二玻片约呈30—45度的角度平稳地向前推至玻片另一端。推时角度要一致,用力应均匀,即推出均匀的血膜(血膜不可过厚、过薄)。将制好的血涂片晾干,不可加热。
注:(1)良好涂片标准:
1) 呈头、体、尾舌形
2) 血膜厚薄适宜
3) 两边留有空隙 (2)涂片好坏与下列因素有关: 1)血滴大小 2)推片与玻片之间角度 3)推片速度
2、血涂片的染色步骤:
(1)用蜡笔在血膜两端各划一道线,以免染料外溢,置涂片于染色架上。
(2)滴加瑞氏染液,共计七八滴数,以盖满血膜为度,静置1分钟。
(3)再滴加等量的磷酸缓冲盐溶液,轻轻摇动,并轻吹液体使染色液与缓冲液混合均匀,静置15分钟。
(4)用清水冲洗。切勿先倾去染液再冲洗,否则沉淀物附于血膜上不宜出去。冲洗后斜置血涂片于空气中干燥。或先用滤纸吸取水分迅速干燥,即可镜检。
3、白细胞分类计数:先用低倍镜检查涂片及染色是否均匀。然后加一滴香柏油于血膜厚薄均匀处(一般在体尾交界处),在油镜下由此处开始按其形态特征进行分类计数,计数移动时避免重复。根据所见到的100个白细胞,记录各种白细胞所占的百分数。
4、各类白细胞的形态特征
嗜中性白粒细胞:圆形,胞浆淡红色,胞浆内有多量紫红色细小颗粒(染色不佳时则看不清楚)。核为蓝紫色。
嗜酸性白细胞:大小、形态与中性白细胞大体相似,唯胞浆内含有粗大的鲜红色颗粒。核多数为两叶。
嗜碱性白细胞:大小、形态与中性白细胞大体相似,但胞浆内含有粗大的深蓝色,常遮盖在核上。核分叶不明显
淋巴细胞:分大小两种,小淋巴细胞圆形,核染成深紫色,圆形或肾形,胞浆很少,常呈一小片月牙状,染成蓝色。大淋巴细胞,核圆形或肾形,胞浆相对较多,染成天蓝色,在胞浆与核之间有淡染带。有时内含少数紫红色大颗粒。
单核细胞: 核染成紫色,其染色质疏松,核形不规则。胞浆较多,染成浅灰蓝色
白细胞计数
加稀释液 用吸管吸取白细胞稀释液0.38ml于小试管中。
吸取血液 用微量吸管吸取新鲜全血或末梢血20ul,擦去管尖外部余血。将吸管插入小试管中白细胞稀释液的底部,轻轻放出血液,并吸取上层白细胞稀释液清洗吸管2-3次
混匀
冲池 将试管中血液与稀释液混匀,待细胞悬液完全变为棕褐色 再次将小试管中的细胞悬液混匀。用滴棒蘸取细胞悬液1滴,充入改良Neubauer计数板的计数池中,室温静置2-3min,待白细胞完全下沉。
计数 在低倍镜下计数四角4个大方格内的白细胞总数
计算 白细胞总数 =4大格白细胞数(N) /4×10 ×20 ×10^6
= N÷ 20 ×10^9/L
注:计数板上下各一个计数池计数区域分为9个大方格,四角的大方格各划分为16个中方格,中央大方格划分为25个中方格。四角的大方格为白细胞计数区,大方格四边压线细胞计左不数右,计上不数下
实验结果
白细胞总数 白细胞=(35+42+39+37) /20*10^9=7.65*10^9/L
中性粒细胞65个,占65% 单核细胞5个,占5%
淋巴细胞:27个,占27% 嗜酸性粒细胞3个,占2%
嗜碱性粒细胞1个,占1%
注意事项
1、.采血时针刺深度必须适当,不能过度挤压,防止组织液渗入或采血时间太长引起血液凝固。
2、白细胞总数在参考范围内,大方格间的细胞数不得相差10个以上,两次重复计数误差不得超过10%。
3、按一定顺序移动视野。
4、染色时切勿使染液干涸,否则发生不易去掉的沉淀。
5、水冲洗时间不宜过长,否则会脱色。
6、血涂片不可过厚
思考题
1.白细胞核不着色可能是因为操作中那种误差引起的?
第二篇:细胞计数方法------细胞计数板法
 细胞计数方法------细胞计数板法
细胞计数方法------细胞计数板法
实验原理:当待测细胞悬液中细胞均匀分布时,通过测定一定体积悬液中的细胞的数目,即可换算出每毫升细胞悬液中细胞的细胞数目。
具体操作:
1. 将计数板及盖片擦拭干净,并将盖片盖在计数板。
2. 将细胞悬液吸出少许,滴加在盖片边缘,使悬液充满盖片和计数板之间,静置3min,注意盖片下不要有气泡,也不能让悬液流入旁边槽中。
3. 计算板四大格细胞总数,压线细胞只计左侧和上方的。然后按公式计算:
细胞数/mL=四大格细胞总数/4×10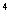 个/ml
个/ml
(注:当细胞很多时,可在四个格中选一定数目较平均的小格,由于每大格中有16个小格,然后计左侧和上方的细胞数,求出每小格的细胞数,取平均值m,m×16即每个格的平均值。所以,细胞密度=m×16×10个/ml)
说明:公式中除以4,因为计数了4个大格的细胞数。
公式中乘以10因为计数板中每一个大格的体积为:
1.0mm(长)×1.0mm(宽)×0.1mm(高)=0.1mm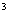 而 1ml=1000ul=1000mm
而 1ml=1000ul=1000mm
(注意:镜下偶见有两个以上细胞组成的细胞团,应按单个细胞计算,若细胞团10%以上,说明分散不好,需重新制备细胞悬液。)
================================================
细胞计数板的使用
一、血球计数板-基本构造
血球计数板是一块特制的厚型载玻片,载玻片上有四个槽构成三个平台。中间的平台较宽,其中间又被一短横槽分隔成两半,每个半边上面各刻有一小方格网,每个方格网共分九个大格,中央的一大格作为计数用,称为计数区。计数区的刻度有两种:一种是计数区分为16个大方格(大方格用三线隔开),而每个大方格又分成25个小方格;另一种是一个计数区分成25个大方格(大方格之间用双线分开),而每个大方格又分成16个小方格。但是不管计数区是哪一种构造,它们都有一个共同特点,即计数区都由400个小方格组成。
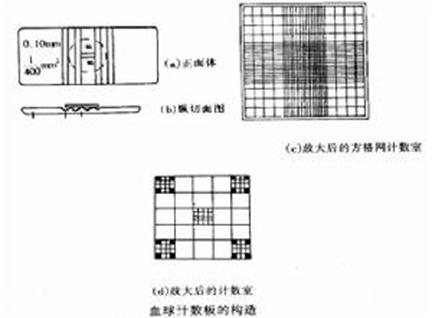
计数区边长为1mm,则计数区的面积为1mm2,每个小方格的面积为1/400mm2。盖上盖玻片后,计数区的高度为0.1mm,所以每个计数区的体积为0.1mm3,每个小方格的体积为1/4000mm3。
使用细胞计数板计数时,先要测定每个小方格中微生物的数量,再换算成每毫升菌液(或每克样品)中微生物细胞的数量。
二、细胞计数板-使用方法
1.视待测菌悬液浓度,加无菌水适当稀释(斜面一般稀释100倍),以每小格的菌数可数为度。
2.取洁净的细胞计数板一块,在计数区上盖上一块盖玻片。
3.将菌悬液摇匀,用滴管吸取少许,从计数板中间平台两侧的沟槽内沿盖玻片的下边缘摘入一小滴(不宜过多),让菌悬液利用液体的表面张力充满计数区,勿使气泡产生,并用吸水纸吸去沟槽中流出的多余菌悬液。也可以将菌悬液直接滴加在计数区上(不要使计数区两边平台沾上菌悬液,以免加盖盖玻片后,造成计数区深度的升高),然后加盖盖玻片(勿使产生气泡)。
4.静置片刻,使细胞沉降到计数板上,不再随液体漂移。将细胞计数板放置于显微镜的载物台上夹稳,先在低倍镜下找到计数区后,再转换高倍镜观察并计数。由于生活细胞的折光率和水的折光率相近,观察时应减弱光照的强度。
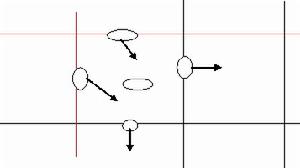
5.计数时若计数区是由16个大方格组成,按对角线方位,数左上、左下、右上、右下的4个大方格(即100小格)的菌数。如果是25个大方格组成的计数区,除数上述四个大方格外,还需数中央1个大方格的菌数(即80个小格)。为了保证计数的准确性,避免重复计数和漏记,在计数时,对沉降在格线上的细胞的统计应有统一的规定。如菌体位于大方格的双线上,计数时则数上线不数下线,数左线不数右线,以减少误差。即位于本格上线和左线上的细胞计入本格,本格的下线和右线上的细胞按规定计入相应的格中。见下图:即本格中计数细胞为3个。
6.对于出芽的酵母菌,芽体达到母细胞大小一半时,即可作为两个菌体计算。每个样品重复计数2-3次(每次数值不应相差过大,否则应重新操作),按公式计算出每mL(g)菌悬液所含细胞数量。
7.测数完毕,取下盖玻片,用水将细胞计数板冲洗干净,切勿用硬物洗刷或抹擦,以免损坏网格刻度。洗净后自行晾干或用吹风机吹干,放入盒内保存。
三、细胞计数板-计数公式
1、16格×25格的细胞计数板计算公式:细胞数/ml=100小格内细胞个数/100×400×10000×稀释倍数
1、25格×16格的细胞计数板计算公式:细胞数/ml=80小格内细胞个数/80×400×10000×稀释倍数
四、血细胞计数板计数的误差主要来自哪些方面?应如何尽量减少误差,力求准确?
血细胞计数的误差分别来源于技术误差和固有误差。其中由于操作人员采血不顺利,器材处理、使用不当,稀释不准确,细胞识别错误等因素所造成的误差属技术误差;而由于仪器(计数板、盖片、吸管等)不够准确与精密带来的误差称仪器误差,由于细胞分布不均匀等因素带来的细胞计数误差属于分布误差或计数域误差(filed error)。仪器误差和分布误差统称为固有误差或系统误差。技术误差和仪器误差可通过规范操作、提高熟练程度和校正仪器而避免或纠正,但细胞分布误差却难于彻底消除。因此,搞好红细胞计数的质量控制一般需采用以下措施。
1.避免技术误差,纠正仪器误差
(1)所用器材均应清洁干燥,计数板、血盖片、微量吸管及刻度吸管的规格应符合要求或经过校正。①计数板的鉴定:要求计数室的台面光滑、透明,划线清晰,计数室划线面积准确。必要时采用严格校正的目镜测微计测量计数室的边长与底面积,用微米千分尺测量计数室的深度。美国国家标准局(NBS)规定每个大方格边长的误差应小于1%,即1±0.01mm,深度误差应小于2%,即0.1±0.002mm。若超过上述标准,应弃之不用。②血盖片应具有一定的重量,平整、光滑、无裂痕,厚薄均匀一致,可使用卡尺多点测量(至少在9个点),不均匀度在0.002mm之内。必要时采用平面平行仪进行测量与评价,要求呈现密集平行的直线干涉条纹。最简单的评价方法是将洁净的盖片紧贴于干燥的平面玻璃上,若能吸附一定的时间不脱落,落下时呈弧线形旋转,表示盖片平整、厚薄均匀。同时,合格的盖片放置在计数室表面后,与支持柱紧密接触的部位可见到彩虹。精选出的盖片与其他盖片紧密重合后,在掠射光线下观察,如见到完整平行的彩虹条纹表示另一枚盖片质量也符合要求。③目前临床实验室多采用一次性微量采血管采集毛细血管血,除注意定点购买使用信誉较好厂家的产品外,还应对每一批量的采血管进行抽样检查,可通过水银称重法或有色溶液比色法进行校正,误差不应超过±1%。
(2)红细胞稀释液应等渗、新鲜、无杂质微粒。
(3)严格操作,从消毒、采血、稀释、充池到计数都应规范,尤其应注意的是血样稀释及充池时既要作到充分混匀,又要防止剧烈震荡为破坏红细胞。必须一次性充满计数室,防止产生气泡,充入细胞悬液的量以不超过计数室台面与血盖片之间的矩形边缘为宜。
(4)报告法定计量单位。
2.缩小计数域误差或分布误差 由于血细胞在充入计数室后呈随机分布或称Poisson分布( 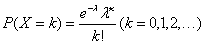 ),而我们所能计数的细胞分布范围是有限的,由此造成的计数误差称为计数域误差或分布误差。缩小这种误差的有效方法就是尽量扩大细胞计数范围和计数数目,一般先进行误差估计,然后决定所需计数的数目和计数范围,只要能将误差控制在允许范围内即可。Berkson指出,当使用同一支吸管、同一面计数室,计数0.2mm2面积的细胞数,有望将 CV控制在可接受的7%以内。对于红细胞计数而言,由于红细胞数量较多,在计数室中显得比较“拥挤”,根据Poisson公式推断, 。欲将误差控制在变异百分数5%以内,至少需要在计数室中计数400个红细胞,因此要求计数五个中方格的红细胞。事实上Berkson还通过实验证明,红细胞的计数域误差为s=0.92 ,较理论误差(Poisson分布误差)要小。
),而我们所能计数的细胞分布范围是有限的,由此造成的计数误差称为计数域误差或分布误差。缩小这种误差的有效方法就是尽量扩大细胞计数范围和计数数目,一般先进行误差估计,然后决定所需计数的数目和计数范围,只要能将误差控制在允许范围内即可。Berkson指出,当使用同一支吸管、同一面计数室,计数0.2mm2面积的细胞数,有望将 CV控制在可接受的7%以内。对于红细胞计数而言,由于红细胞数量较多,在计数室中显得比较“拥挤”,根据Poisson公式推断, 。欲将误差控制在变异百分数5%以内,至少需要在计数室中计数400个红细胞,因此要求计数五个中方格的红细胞。事实上Berkson还通过实验证明,红细胞的计数域误差为s=0.92 ,较理论误差(Poisson分布误差)要小。
3.排除异常标本的干扰 白细胞数量在正常范围时,相对于红细胞数量来讲,其影响可忽略,但如白细胞过高(>100×109/L),则应对计数结果进行校正。方法是:①实际RBC=计得RBC-WBC。如当红细胞换算后为3.5×1012/L、白细胞换算后为100×109/L时,病人实际红细胞数应为3.4×1012/L。②在高倍镜下计数时,避开有核细胞。有核细胞体积比正常红细胞大,中央无凹陷,无草黄色折光,可隐约见到细胞核。此外,当病人急性严重贫血时网织红细胞可提前大量释放,也给红细胞计数带来一定的干扰,而且影响网织红细胞绝对值计算结果。其校正方法有待探讨。
Using a Counting Chamber
For microbiology, cell culture, and many applications that require use of suspensions of cells it is necessary to determine cell concentration. One can often determine cell density of a suspension spectrophotometrically, however that form of determination does not allow an assessment of cell viability, nor can one distinguish cell types.
A device used for determining the number of cells per unit volume of a suspension is called a counting chamber. The most widely used type of chamber is called a hemocytometer, since it was originally designed for performing blood cell counts.
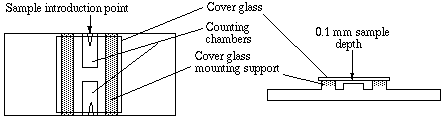
To prepare the counting chamber the mirror-like polished surface is carefully cleaned with lens paper. The coverslip is also cleaned. Coverslips for counting chambers are specially made and are thicker than those for conventional microscopy, since they must be heavy enough to overcome the surface tension of a drop of liquid. The coverslip is placed over the counting surface prior to putting on the cell suspension. The suspension is introduced into one of the V-shaped wells with a pasteur or other type of pipet. The area under the coverslip fills by capillary action. Enough liquid should be introduced so that the mirrored surface is just covered. The charged counting chamber is then placed on the microscope stage and the counting grid is brought into focus at low power.
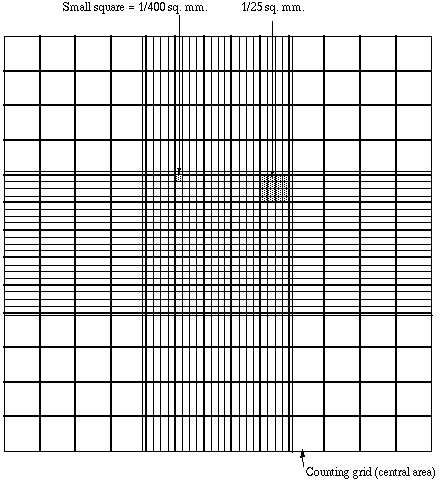
It is essential to be extremely careful with higher power objectives, since the counting chamber is much thicker than a conventional slide. The chamber or an objective lens may be damaged if the user is not not careful. One entire grid on standard hemacytometers with Neubauer rulings can be seen at 40x (4x objective). The main divisions separate the grid into 9 large squares (like a tic-tac-toe grid). Each square has a surface area of one square mm, and the depth of the chamber is 0.1 mm. Thus the entire counting grid lies under a volume of 0.9 mm-cubed
Suspensions should be dilute enough so that the cells or other particles do not overlap each other on the grid, and should be uniformly distributed. To perform the count, determine the magnification needed to recognize the desired cell type. Now systematically count the cells in selected squares so that the total count is 100 cells or so (number of cells needed for a statistically significant count). For large cells this may mean counting the four large corner squares and the middle one. For a dense suspension of small cells you may wish to count the cells in the four 1/25 sq. mm corners plus the middle square in the central square. Always decide on a specific counting patter to avoid bias. For cells that overlap a ruling, count a cell as "in" if it overlaps the top or right ruling, and "out" if it overlaps the bottom or left ruling.
Here is a way to determine a particle count using a Neubauer hemocytometer. Suppose that you conduct a count as described above, and count 187 particles in the five small squares described. Each square has an area of 1/25 mm-squared (that is, 0.04 mm-squared) and depth of 0.1 mm. The total volume in each square is (0.04)x(0.1) = 0.004 mm-cubed. You have five squares with combined volume of 5x(0.004) = 0.02 mm-cubed. Thus you counted 187 particles in a volume of 0.02 mm-cubed, giving you 187/(0.02) = 9350 particles per mm-cubed. There are 1000 cubic millimeters in one cubic centimeter (same as a milliliter), so your particle count is 9,350,000 per ml.
Cells are often large enough to require counting over a larger surface area. For example, you might count the total number of cells in the four large corner squares plus the middle combined. Each square has surface area of 1 mm-squared and a depth of 0.1 mm, giving it a volume of 0.1 mm-cubed. Suppose that you counted 125 cells (total) in the five squares. You then have 125 cells per 0.5 mm-cubed, which is 250 cells/mm-cubed. Again, multiply by 1000 to determine cell count per ml (250,000).
Sometimes you will need to dilute a cell suspension to get the cell density low enough for counting. In that case you will need to multiply your final count by the dilution factor. For example, suppose that for counting you had to dilute a suspension of Chlamydomonas 10 fold. Suppose you obtained a final count of 250,000 cells/ml as described above. Then the count in the original (undiluted) suspension is 10 x 250,000 which is 2,500,000 cells/ml.
-
细胞计数实验报告
细胞计数实验报告一目的培养的细胞在一般条件下要求有一定的密度才能生长良好所以要进行细胞计数二原理细胞计数的原理和方法与血细胞计数相…
-
实验 白细胞计数和分类计数
白细胞计数和分类计数第一临床医学院08中七2班董盼攀20xx1150226一实验目的11掌握显微镜法白细胞计数的原理及方法数的方法…
-
白细胞计数实验报告
白细胞计数和分类目的掌握血涂片制备的操作要领瑞氏染色方法正常外周血五种白细胞形态特点原理各种白细胞必须经过染色才易于区分其类别常用…
-
红、白细胞计数实验报告
实验诊断学实验报告实验一红细胞白细胞计数一实验原理一定量的血液经一定量等渗性稀释液稀释后充入血细胞计数池中于显微镜下计数一定体积内…
-
红细胞计数、白细胞计数实验报告
一实验原理用等渗稀释液将血液稀释一定倍数充入计数池后在显微镜下计数一定体积内的红细胞数量经换算求出每升血液中的红细胞数量用白细胞稀…
-
实验 白细胞计数和分类计数
白细胞计数和分类计数第一临床医学院08中七2班董盼攀20xx1150226一实验目的11掌握显微镜法白细胞计数的原理及方法数的方法…
-
红、白细胞计数实验报告
实验诊断学实验报告实验一红细胞白细胞计数一实验原理一定量的血液经一定量等渗性稀释液稀释后充入血细胞计数池中于显微镜下计数一定体积内…
-
红细胞计数、白细胞计数实验报告
一实验原理用等渗稀释液将血液稀释一定倍数充入计数池后在显微镜下计数一定体积内的红细胞数量经换算求出每升血液中的红细胞数量用白细胞稀…
-
细胞生物学综合性实验报告
本科学生实验报告学号姓名学院生命科学学院专业班级11级生物科学C班实验课程名称细胞生物学实验指导教师及职称吴暇玉开课时间20xx至…
-
细胞工程实验报告
细胞工程实验报告专业生物技术班级0801姓名励丹学号30804305一实验目的1了解动物细胞培养的相关实验器材以及实验前器材的清洗…
-
《实验诊断学》红细胞与白细胞数量测定实验
实验诊断学实验报告实验一红细胞计数白细胞计数学院专业年级学号姓名实验日期20xx一实验原理1红细胞计数用等渗稀释液将血液稀释一定倍…