气相色谱实验
气相色谱法测定混合物中环己烷含量
一、实验的目的和要求
1.掌握气相色谱仪的基本构成及基本操作;
2.掌握气相色谱柱分离化学物质的原理;
3.掌握气相色谱中保留值定性与内标法定量的分析方法。
二、实验原理
(一)气相色谱分离原理
气相色谱仪是一种多组份混合物的分离、分析工具,它是以气体为流动相,采用冲洗法的柱色谱技术。当多组份的分析物质进入到色谱柱时,由于各组分在色谱柱中的气相和固定液液相间的分配系数不同,当气化后的试样被载气带入色谱柱中运行时,组份就在其中的两相间进行反复多次的分配(吸附-脱附或溶解-释放),由于固定相对各组份的吸附或溶解能力不同(即保留作用不同),因此各组份在色谱柱中的运行速度就不同,经过一定的柱长后,便彼此分离,顺序离开色谱柱进入检测器,经检测后转换为电信号送至色谱数据处理装置处理,从而完成了对被测物质的定性定量分析。
气相色谱法特点:(1)分离分析连贯性强,方法简便;(2)分离效能高,选择性好;(3)分析速度快;(4)灵敏度高;(5)应用范围广。
(二)气相色谱仪器组成
气相色谱仪一般由五部分组成:
(1) 载气系统,包括气源、气体净化、气体流速控制和测量。
(2) 进样系统,包括进样口、气化室。
(3) 色谱柱和柱箱,包括恒温控制装置。
(4) 检测系统,包括检测器及控温装置。
(5) 记录系统,包括放大器、记录仪,有的仪器还有数据处理装置。
气相色谱仪的工作流程如图1所示。载气由高压钢瓶供给,经减压阀减压后,进入气路控制单元控制载气的压力和流量,再经过进样口(包括气化室),试样就在进样口注入(如为液体试样,经气化室瞬间气化为气体)。由载气携带进入色谱柱,将各组分分离后依次进入检测器后放空。检测器信号由数据处理装置记录,就可得到色谱图。
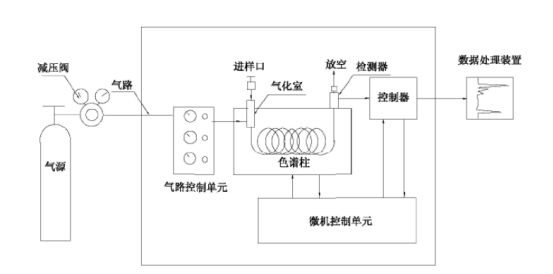
图1 气相色谱流程图
(三)气相色谱的分析方法
1、样品的定性
用纯物质的保留值对照定性。在一个确定的色谱条件下,每一个物质都有一个确定的保留值,所以在相同条件下,未知物的保留值和已知物的保留值相同时,就可以认为未知物即是用于对照的已知纯物质。但是,有不少物质在同一条件下可能有非常相近的而不容易察觉差异的保留值,所以,当样品组分未知时,仅用纯物质的保留值与样品组分的保留值对照定性是困难的。这种情况,需用两根不同的极性的柱子或两种以上不同极性固定液配成的柱子,对于一些组成基本上可以估计的样品,那么准备这样一些纯物质,在同样的色谱条件下,以纯物质的保留时间对照,用来判断其色谱峰属于什么组分是一种简单而行方便的定性方法。
用标准加入法来定性。首先用未知的混合样品在一定的色谱条件下采集混合物样品的色谱峰,然后取一定量的混合物样品中加入怀疑有的物质的纯物质,在相同的色谱条件下采集加入某纯物质的色谱峰,用两个色谱图进行比较,就会发现两个色谱图上某一个峰的保留值相同,但加了某纯物质的色谱图上的色谱峰的峰高增加、峰面积增大,那么此峰即为某纯物质。
2.样品的定量
(1)色谱定量分析的依据
在一定操作条件下,分析组分i的重量(mi)或其在载气中的浓度是与检测器的响应信号(峰高或峰面积)成正比的,可写作:

这是色谱定量分析的依据。由此可见,定量分析中需要:(1)准确测量峰面积;(2)准确求出比例系数f(定量校正因子);(3)正确选用定量计算方法,将测得组分的峰面积换算为百分含量。
(2)校正因子的测量
校正因子有绝对校正因子和相对校正因子。
绝对校正因子 是指
是指 物质进样量
物质进样量 与它的峰面积
与它的峰面积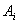 或峰高
或峰高 之比:
之比:
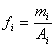 或
或 
只有在仪器条件和操作条件严格恒定的情况下,一种物质的绝对校正因子才是稳定值,才有意义。同时,要准确测定绝对校正因子,还要求有纯物质,并能准确知道进样量,所以它的应用受到限制。
相对校正因子是指物质的绝对校正因子与作为基准的 物质的绝对校正因子之比。可以表示为:
物质的绝对校正因子之比。可以表示为:
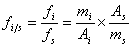
测定相对校正因子,只需配制和的质量比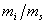 为已知的标样,进样后测出它们的峰面积之比
为已知的标样,进样后测出它们的峰面积之比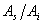 ,即可计算出
,即可计算出 。进样多少,不必准确计量,所以相对校正因子更容易测定。而且,只要是同类检测器,色谱条件不同时,相对校正因子基本上保持恒定,使用中不必要操作条件严格相同,适应性和通用性更强。
。进样多少,不必准确计量,所以相对校正因子更容易测定。而且,只要是同类检测器,色谱条件不同时,相对校正因子基本上保持恒定,使用中不必要操作条件严格相同,适应性和通用性更强。
(3)归一化法定量
归一化法定量的依据是,当样品的所有组分均出峰时,那么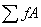 就代表了样品的进样量,其某一部分的进样量则为
就代表了样品的进样量,其某一部分的进样量则为 ,组分的百分率为:
,组分的百分率为:

所以归一化法测定时,就是在测定组分的相对校正因子后,将样品中所有组分的峰面积测出,按此式计算各组分的百分含量。
(4)内标法
将一定量的纯物质作为内标物,加入到准确称取的样品中,根据被测物的重量及其在色谱图上相应的峰面积比,求出某组分的含量,设mi、ms分别为被测物和内标物的重量,则mi=fiAi,ms=fsAs,mi/ms= fiAi/ fsAs,则:


一般以内标物为基准,则fs=1,此时计算式可简化为:

优点:定量较准确,适用于所有组分不能全部出峰的情况。
(5)外标法
用欲测组分的纯物质来制作标准曲线,以响应信号为纵坐标,以百分含量为横坐标绘制标准曲线,分析试样时进样量与绘制曲线时进样量相同。经色谱分析测得该样品的响应信号(如峰面积或峰高),再由所制的标准曲线上查得相应的含量值。
优点:操作简单,计算方便,但结果的准确度主要取决于进样量的重现性和操作条件的稳定性。
本实验采用内标法完成样品定量分析。
三、实验仪器与试剂
1.仪器:SP-2100A气相色谱仪;FID检测器;N2000双通道色谱工作站;HP-5毛细管色谱柱;微量进样器。
2.试剂:正己烷(分析纯),环己烷(分析纯),无水乙醇(分析纯),庚烷(分析纯)。
四、实验步骤
1. 样品的配制:称取3.00克正己烷,2.00克环己烷,用无水乙醇定容到50ml,称量样品总重量。准确称取3.00克庚烷(内标),加入到已称重的待测样品中,混合均匀供实验用。
2. 内标物和组分定性分析:
(1) 取环己烷和庚烷(内标)分别进样进行气相色谱测定,记录其保留时间和峰面积。
(2) 取配好的样品在相同的色谱条件下进样测定,分别记录各谱峰的保留时间。
(3) 将(1)和(2)中测定的保留时间相对照,确定样品中环己烷和庚烷(内标)在谱图中的位置。
3. 校正因子的测定:
利用步骤2中(1)测出的峰面积计算环己烷的校正因子:
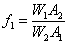
4. 样品中环己烷的定量测定:
取实验样品进样做气相色谱测定,记录环己烷和内标物庚烷的峰面积。
5. 仪器操作条件:
检测器:FID
柱温:100℃
进样口温度:200℃
检测器温度:200℃
载气流速:30ml/min
进样量:1μl。
6.气相色谱仪操作步骤:
(1)打开载体钢瓶总阀,观察载气压力是否到达预定值。
(2)开启主机电源,设置相应参数。
(3)打开计算机电源,启动色谱工作站。
(4)打开氢气发生器开关,观察压力是否显示至预定值。
(5)打开空气压缩机开关,观察压力是否显示至预定值。
(6)点燃FID火焰。
(7)待主机显示“就绪”,观察记录仪信号,待基线平稳后,开始测试。
(8) 测试完毕后,先在主机上设置进样器、柱温和检测器温度至50℃,当进样器、柱温和检测器温度降至60℃以下时,关闭主机电源,退出色谱工作站并关闭电源,关闭氢气发生器、空气压缩机开关,关闭载气钢瓶总阀。
五、数据处理
将实验中测得的内标物庚烷和样品环己烷校正因子、峰面积以及内标物、混合样品的量代入内标法定量计算公式中,计算出样品中环己烷的百分含量。
六、实验报告要求
写清楚实验的目的和原理、实验操作步骤以及实验条件,并正确处理实验数据。
七、思考题
1. 在内标法定量分析中,内标物的选择是很重要的,你知道内标物的选择原则吗?
2.内标物与被测样品含量的比例多少比较合适?
3.什么时候用归一化法?
第二篇:气相色谱定性分析
华南师范大学实验报告
专 业: 材料化学 年 级:20##级
课程名字:近代材料分析测试技术 实验项目:气相色谱定性分析
实验类型: 验证 实验时间:20##年3月4日
实验一 气相色谱定性分析
一、目的要求
1、 学习利用纯物对照法和加入纯物增加峰高法的定性方法;
2、 熟悉色谱仪器操作。
二、基本原理
在一定的色谱条件下,一个未知物只有一个确定的保留时间。因此,对于较简单的多组分混合物,若其中所有待测组分均为已知,它们的色谱峰均能分开,则可将已知纯物质在相同的色谱条件下的保留时间与未知物的保留时间进行比较,就可以定性鉴定未知物。纯物质对照法定性只适用于组分性质已有所了解,组成比较简单,且有纯物质的未知物。
当未知样品中组分较多,所得色谱峰过密,用上述方法不易辨认时,或仅作未知样品指定项目分析时均可用此法。首先做出未知样品的色谱图,然后在未知样品加入某已知物,又得到一个色谱图。峰高增加的组分即可能为这种已知物。
三、仪器及设备
GC-2010气相色谱仪;SHIMADZU-日本岛津;色谱工作站;
色谱柱:开管柱,长30m,中等极性,内径:0.32mm,膜厚:0.25μm。
全自动空气源,空气压缩机,氢气发生器;1uL微量进样器。
四、实验试剂:
甲苯(分析纯)、苯乙烯,未知样品,请选作。
五、实验条件
1、温度:进样温度150℃;柱温80℃左右;检测器温度150℃。
2、气体流量:载气为氮气 40mL/min,空气400 mL/min,氢气40mL/min。
3、检测器 FID,灵敏度10-7。
4、进样量 0.2uL
六、实验步骤
1、 色谱仪器进样操作;
2、 纯物对照法:
1)进标样:分别吸取甲苯、苯乙烯各0.2uL,依次进样,准确记录保留时间。
2)进待测样:用待测样把1uL微量进样器洗3-5次,然后往色谱仪内注射0.2 uL样品,准确记录保留时间。
3)将甲苯、苯乙烯标样的保留时间与待测样的保留时间对比定性。
3、 加入纯物增加峰高:
1)进待测样:用待测样把1uL微量进样器洗3-5次,然后往色谱仪内注射0.2 uL样品,准确记录保留时间。
2)取上述待测样二份,分别加入适量甲苯、苯乙烯标样,分别吸取配制的混合样品0.2uL,依次进样,观察色谱峰变化。
3)根据色谱峰峰高变化定性。
七、结果处理:
标出各峰的保留时间等,计算分离度,对不同组分计算色谱柱的分离效能(计算塔板数)。
(一)记录样品各峰保留时间,如下表:

(二)求分离度
已知半峰宽Y1/2= =2.354σ 峰基宽W = 4 σ 所以W = 1.699Y1/2
=2.354σ 峰基宽W = 4 σ 所以W = 1.699Y1/2
则可求出分离度 R= 
根据实验实验所得各样品色谱峰图片,测量并计算半峰宽Y1/2,再算出峰基宽W,从而可算出样品分离度R。如下表

(三)计算色谱柱的分离效能
所以 n = 

(四)计算混合物的比例
已知ρ甲苯 = 0.866 g/mol ;ρ苯乙烯 = 0.906 g/mol
在体积比为1:1的甲苯和苯乙烯混合溶液中,V甲苯=V苯乙烯=0.2 uL
求得甲苯的质量分数X1为:
X1 = 
= 
=( 0.866 x 0.1 ) / (0.866 x 0.1 + 0.906 x 0.1)
= 48.87%
用归一化法求解校对因子,设甲苯和苯乙烯的校对因子分别为f1、f2, 甲苯和苯乙烯的峰面积分别A、B
由实验得A= 574000215.7 * 10^7 B= 642279560.6 * 10^7
X1= 
由上式可得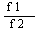 =
= 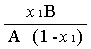 = 1.07
= 1.07
不妨设f2 = 1 ,则f1= 1.07
在未知比例的混合液中,由实验得A1=409404060.6 * 10^7 μV B1=754237355.6 * 10^7 μV
则 X2= 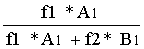 = 34.16%
= 34.16%
则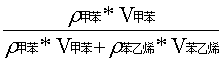 =34.16% 可求得
=34.16% 可求得 =1.84
=1.84
所以混合物甲苯和苯乙烯之比为:1:1.84
八、结果讨论
有很多因素影响柱效,如柱温、载气流量、固定液含量、载气性质、进样速度、进样量等,以下根据实验结果进行分析。
(一) 分子扩散因素、其它因素对柱内传质的影响
由速率理论探讨影响柱效的动力学因素。柱效可用板高H来表示,板高H越小,柱效越大。
由Van Deemter方程,得:

u :流动相线速度; A,B,C为常数,
A—表示涡流扩散系数
B—分子扩散系数
C—传质阻力系数(包括液相和固相传质阻力系数)
1)涡流扩散项(Multipath term, A)
在填充柱中,由于受到固定相颗粒的阻碍,组份在迁移过程中随流动相不断改变方向,形成紊乱的“涡流”。因填充物颗粒大小及填充的不均匀性影响同一组分运行路线,从而影响流出时间,导致峰形展宽。展宽程度以A表示:
A=2ldp
(其中dp—填充物平均直径; l—填充不规则因子。)
所以使用细粒的固定相并填充均匀可减小A,提高柱效。
2)分子扩散项(Longitudinal diffusion term, B/u)
纵向分子扩散是由于浓度梯度引起的。当样品被注入色谱柱时,由于溶质区带前后存在着浓度差,因而产生纵向扩散使色谱峰扩宽。其大小为:
B=2gD?
(g—弯曲因子,指固定相几何形状对自由分子扩散的阻碍情况;D—组分的扩散系数。)
? 分子量大的组分,Dg小,即B小;
? Dg 随柱温升高而增加,随柱压降低而减小;
? 流动相分子量大,Dg小,即B小;
? u 增加,组份停留时间短,纵向扩散小(B/u)。
所以大分子量流动相;适当增加流速;短柱;低温,可提高柱效。
3)传质阻力项(Mass-transfer term, Cu)
? 因传质阻力的存在,使分配不能“瞬间”达至平衡,因此产生峰形展宽。气相色谱以气体为流动相,液相色谱以液体为流动相,二者传质过程不完全相同。
 ? 传质阻力项包括固定相传质阻力项、流动相传质阻力项。在流速很高的情况下,由于没有足够的时间建立平衡,偏离更为严重。气相色谱分析法中,固定填充柱,对某一物质在不同的载气流速下,测定塔板高度,即可作右图。
? 传质阻力项包括固定相传质阻力项、流动相传质阻力项。在流速很高的情况下,由于没有足够的时间建立平衡,偏离更为严重。气相色谱分析法中,固定填充柱,对某一物质在不同的载气流速下,测定塔板高度,即可作右图。
? 从图中可以看出,曲线最低点处 H 最小,即柱效能最高,该点对应的载气流速为最佳流速。
(二)柱外效应
柱外效应:简单的说色谱峰在柱外死空间里的扩展效应。柱外效应除了由不好的进样技术引起外;主要是由从进样点到检测池之间除柱子以外的所有死体积所引起。死体积通常测试中是包括了柱外死体积,即色谱仪中的管路和连接头间的空间以及进样系统和检测器的空间,包括柱后死体积和柱前死体积:
A)柱后死体积如果太大就会造成已分离物的重新混合,使分离度降低峰形变差;
B)柱前死体积太大造成样品分散过大,其效果类似于增加了上样体积,影响色谱柱的分离效果。
九、参考文献
[1] 杨睿. 周啸. 罗传秋. 汪昆华.聚合物近代仪器分析(第三版). 北京:清华大学出版社,2010.6.
-
气相色谱法实验报告
色谱与光谱实验实验五气相色谱法实验色谱与光谱实验色谱与光谱实验气相色谱法实验一实验目的1了解气相色谱仪的各部件的功能2加深理解气相…
-
气相色谱实验报告
气相色谱实验报告姓名XXX专业有机化学学号3120xx303004时间20xx1026一实验目的1了解气相色谱的仪器组成工作原理及…
-
气相色谱实验报告
气相色谱实验报告一实验目的1了解气相色谱仪的基本结构及掌握分离分析的基本原理2了解顶空气相色谱法3了解影响分离效果的因素4掌握定性…
-
气相色谱实验报告
仪器分析实验报告实习名称学院专业化学工程与工艺班级化工班姓名学号同组者姓名指导教师日期一实验目的1了解气相色谱仪的基本构成及基本操…
-
气相色谱实验
气相色谱法测定混合物中环己烷含量一实验的目的和要求1掌握气相色谱仪的基本构成及基本操作2掌握气相色谱柱分离化学物质的原理3掌握气相…
-
气相色谱法实验报告
色谱与光谱实验实验五气相色谱法实验色谱与光谱实验色谱与光谱实验气相色谱法实验一实验目的1了解气相色谱仪的各部件的功能2加深理解气相…
-
气相色谱实验报告
气相色谱实验报告一实验目的1了解气相色谱仪的基本结构及掌握分离分析的基本原理2了解顶空气相色谱法3了解影响分离效果的因素4掌握定性…
-
气相色谱实验报告lyh
实验日期20##/3/20成绩同组人##闽南师范大学应用化学专业实验报告题目:气相色谱法测定醇醚混合物12应化1##0前言0.1实…
-
分析实验报告 高效液相色谱
华南师范大学实验报告学生姓名:##学号:专业:化学年级班级:08化二课程名称:仪器分析实验实验项目:液相色谱分析混合样品中的苯和甲…
-
液相色谱实验报告
仪器分析实验报告实习名称液相色谱分析实验学院专业化学工程与工艺班级化工班姓名学号指导教师日期一实验目的1了解液相色谱的构造原理及操…
-
色谱法测定固体催化剂的表面积实验报告
实验四色谱法测定固体催化剂的表面积一实验目的1掌握用流动吸附色谱法测定催化剂比表面积的方法2通过实验了解BET多层吸附理论在测定比…