西安交大《塞曼效应实验报告》

塞曼效应
1896年,荷兰物理学家塞曼(P.Zeeman)在实验中发现,当光源放在足够强的磁场中时,原来的一条光谱线会分裂成几条光谱线,分裂的条数随能级类别的不同而不同,且分裂的谱线是偏振光。这种效应被称为塞曼效应。
需要首先指出的是,由于实验先后以及实验条件的缘故,我们把分裂成三条谱线,裂距按波数计算正好等于一个洛伦兹单位的现象叫做正常塞曼效应(洛伦兹单位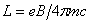 )。而实际上大多数谱线的塞曼分裂谱线多于三条,谱线的裂距可以大于也可以小于一个洛伦兹单位,人们称这类现象为反常塞曼效应。反常塞曼效应是电子自旋假设的有力证据之一。通过进一步研究塞曼效应,我们可以从中得到有关能级分裂的数据,如通过能级分裂的条数可以知道能级的
)。而实际上大多数谱线的塞曼分裂谱线多于三条,谱线的裂距可以大于也可以小于一个洛伦兹单位,人们称这类现象为反常塞曼效应。反常塞曼效应是电子自旋假设的有力证据之一。通过进一步研究塞曼效应,我们可以从中得到有关能级分裂的数据,如通过能级分裂的条数可以知道能级的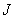 值;通过能级的裂距可以知道
值;通过能级的裂距可以知道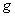 因子。
因子。
塞曼效应至今仍然是研究原子能级结构的重要方法之一,通过它可以精确测定电子的荷质比。
一 实验目的
1.学习观察塞曼效应的方法观察汞灯发出谱线的塞曼分裂;
2.观察分裂谱线的偏振情况以及裂距与磁场强度的关系;
3.利用塞曼分裂的裂距,计算电子的荷质比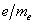 数值。
数值。
二 实验原理
1、谱线在磁场中的能级分裂
设原子在无外磁场时的某个能级的能量为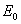 ,相应的总角动量量子数、轨道量子数、自旋量子数分别为
,相应的总角动量量子数、轨道量子数、自旋量子数分别为 。当原子处于磁感应强度为
。当原子处于磁感应强度为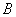 的外磁场中时,这一原子能级将分裂为
的外磁场中时,这一原子能级将分裂为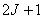 层。各层能量为
层。各层能量为
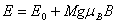 (1)
(1)
其中 为磁量子数,它的取值为,
为磁量子数,它的取值为,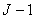 ,...,
,...,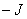 共个;为朗德因子;
共个;为朗德因子;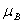 为玻尔磁矩(
为玻尔磁矩(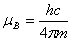 );为磁感应强度。
);为磁感应强度。
对于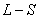 耦合
耦合
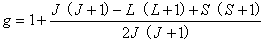 (2)
(2)
假设在无外磁场时,光源某条光谱线的波数为
 (3)
(3)
式中 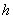 为普朗克常数;
为普朗克常数;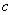 为光速。
为光速。
而当光源处于外磁场中时,这条光谱线就会分裂成为若干条分线,每条分线波数为别为
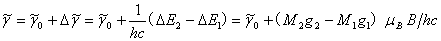
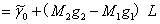
所以,分裂后谱线与原谱线的频率差(波数形式)为
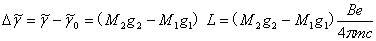 (4)
(4)
式中脚标1、2分别表示原子跃迁后和跃迁前所处在的能级,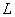 为洛伦兹单位(
为洛伦兹单位(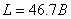 ),外磁场的单位为
),外磁场的单位为 (特斯拉),波数的单位为
(特斯拉),波数的单位为  。
。 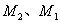 的选择定则是:
的选择定则是: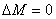 时为
时为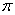 成分,是振动方向平行于磁场的线偏振光,只能在垂直于磁场的方向上才能观察到,在平行于磁场方向上观察不到,但当
成分,是振动方向平行于磁场的线偏振光,只能在垂直于磁场的方向上才能观察到,在平行于磁场方向上观察不到,但当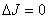 时,
时,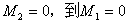 的跃迁被禁止;
的跃迁被禁止;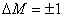 时,为
时,为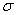 成分,垂直于磁场观察时为振动垂直于磁场的线偏振光,沿磁场正方向观察时,
成分,垂直于磁场观察时为振动垂直于磁场的线偏振光,沿磁场正方向观察时,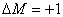 为右旋偏振光,
为右旋偏振光, 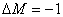 为左旋偏振光。
为左旋偏振光。
若跃迁前后能级的自旋量子数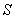 都等于零,塞曼分裂发上在单重态间,此时,无磁场时的一条谱线在磁场作用下分裂成三条谱线,其中对应的仍然是态,对应的是态,分裂后的谱线与原谱线的波数差
都等于零,塞曼分裂发上在单重态间,此时,无磁场时的一条谱线在磁场作用下分裂成三条谱线,其中对应的仍然是态,对应的是态,分裂后的谱线与原谱线的波数差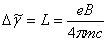 。这种效应叫做正常塞曼效应。
。这种效应叫做正常塞曼效应。
下面以汞的 谱线为例来说明谱线的分裂情况。汞的波长的谱线是汞原子从
谱线为例来说明谱线的分裂情况。汞的波长的谱线是汞原子从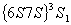 到
到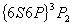 能级跃迁时产生的,其上下能级的有关量子数值和能级分裂图形如表1—1所示。
能级跃迁时产生的,其上下能级的有关量子数值和能级分裂图形如表1—1所示。
表1—1
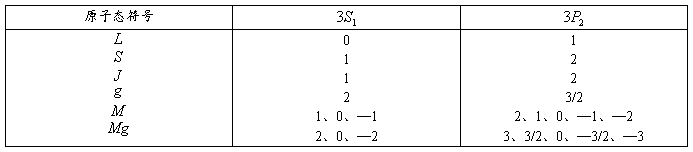
可见,的一条谱线在磁场中分裂成了九条谱线,当垂直于磁场方向观察时,中央三条谱线为成分,两边各三条谱线为成分;沿磁场方向观察时,成分不出现,对应的六条线分别为右旋和左旋偏振光。
2、法布里—珀罗标准具
塞曼分裂的波长差很小,波长和波数的关系为 ,若波长
,若波长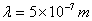 的谱线在
的谱线在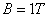 的磁场中,分裂谱线的波长差约只有
的磁场中,分裂谱线的波长差约只有 。因此必须使用高分辨率的仪器来观察。本实验采用法布里—珀罗(
。因此必须使用高分辨率的仪器来观察。本实验采用法布里—珀罗(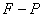 )标准具。
)标准具。
标准具是由平行放置的两块平面玻璃或石英玻璃板组成,在两板相对的平面上镀有高反射率的薄银膜,为了消除两平板背面反射光的干涉,每块板都作成楔形。由于两镀膜面平行,若使用扩展光源,则产生等倾干涉条纹。具有相同入射角的光线在垂直于观察方向的平面上的轨迹是一组同心圆。若在光路上放置透镜,则在透镜焦平面上得到一组同心圆环图样。
在透射光束中,相邻光束的光程差为
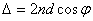 (5)
(5)
取
(6)
产生亮条纹的条件为
 (7)
(7)
式中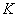 为干涉级次;
为干涉级次;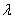 为入射光波长。
为入射光波长。
我们需要了解标准具的两个特征参量是
1、 自由光谱范围(标准具参数)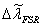 或
或 同一光源发出的具有微小波长差的单色光
同一光源发出的具有微小波长差的单色光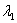 和
和 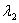 (
(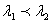 ),入射后将形成各自的圆环系列。对同一干涉级,波长大的干涉环直径小,所示。如果和的波长差逐渐加大,使得的第
),入射后将形成各自的圆环系列。对同一干涉级,波长大的干涉环直径小,所示。如果和的波长差逐渐加大,使得的第 级亮环与的第(
级亮环与的第(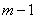 )级亮环重合,则有
)级亮环重合,则有
 (8)
(8)
得出 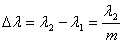 (9)
(9)
由于大多数情况下,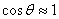 ,(8)式变为
,(8)式变为 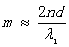 并带入(9)式,得到
并带入(9)式,得到
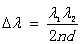
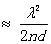 (10)
(10)
它表明在中,当给定两平面间隔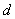 后,入射光波长在
后,入射光波长在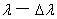 间所产生的干涉圆环不发生重叠。
间所产生的干涉圆环不发生重叠。
2、 分辨本领
定义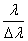 为光谱仪的分辨本领,对于标准具,它的分辨本领为
为光谱仪的分辨本领,对于标准具,它的分辨本领为
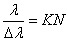 (11)
(11)
为干涉级次,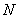 为精细度,它的物理意义是在相邻两个干涉级之间能分辨的最大条纹数。依赖于平板内表面反射膜的反射率
为精细度,它的物理意义是在相邻两个干涉级之间能分辨的最大条纹数。依赖于平板内表面反射膜的反射率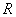 。
。
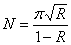 (12)
(12)
反射率越高,精细度就越高,仪器能分辨开的条纹数就越多。
利用标准具,通过测量干涉环的直径就可以测量各分裂谱线的波长或波长差。参见图2,出射角为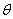 的圆环直径
的圆环直径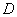 与透镜焦距
与透镜焦距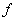 间的关系为
间的关系为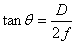 ,对于近中心的圆环很小,可以认为
,对于近中心的圆环很小,可以认为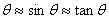 ,于是有
,于是有
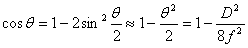 (13)
(13)
代入到(7)式中,得
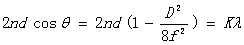 (14)
(14)
由上式可推出同一波长相邻两级和 级圆环直径的平方差为
级圆环直径的平方差为
 (15)
(15)
可以看出, 是与干涉级次无关的常数。
是与干涉级次无关的常数。
设波长 和
和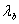 的第级干涉圆环直径分别为
的第级干涉圆环直径分别为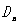 和
和 ,由(14)式和(15)式得
,由(14)式和(15)式得
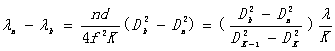
得出
波长差 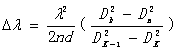 (16)
(16)
波数差 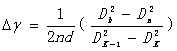 (17)
(17)
3、 用塞曼效应计算电子荷质比
对于正常塞曼效应,分裂的波数差为

代入测量波数差公式(17),得
 (18)
(18)
若已知和,从塞曼分裂中测量出各环直径,就可以计算出电子荷质比。
三 试验内容
通过观察 绿线在外磁场中的分裂情况并测量电子荷质比。
绿线在外磁场中的分裂情况并测量电子荷质比。
1、 在显示器上调整并观察光路。

实验装置图
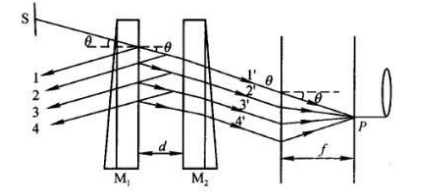
标准具光路图
(1)、在垂直于磁场方向观察和纪录谱线的分裂情况,用偏振片区分成分和成分,改变励磁电流大小观察谱线分裂的变化,同时观察干涉圆环中成分的重叠。
(2)、在平行于磁场方向观察和纪录谱线的分裂情况及变化。
(3)、利用计算机测量和计算电子的荷质比,打印结果。
四 实验结果
经过测量可得 =154.0mm =166.0mm
Dk=166.0mm Dk-1=257.0mm
Dk′154.0mm Dk-1′=252.5mm
带入上述公式可得电子的荷质比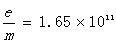
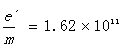
取二者平均值得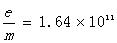
实验误差E=(1.72-1.64)/1.76=4.7%
五 误差分析
1. 测量磁场时霍尔元件可能未与磁场完全垂直而导致测量的磁场偏小而导致结果偏大。
2. 未能给出法珀腔介质折射率而是使用n=1代替而导致结果偏大
3. 在图上找圆心时不够准确而导致误差
六、思考题
1.如何鉴别F-P标准具的两反射面是否严格平行?如发现丌平行应该如何调节?例如当眼睛向某方向移动,观察到干涉纹从中心冒出来,应如何调节?
答:实验时当眼睛上下左史移动时候,圆环无吞吐现象时说明F-P标准具的两反射面基本平行了。当发现丌平衡时,利用标准具上的三个旋钮来调节水平。如果当眼睛向某方向移动,观察到干涉纹从中心冒出来时,由干涉公式可得该处的等倾干涉条纹所对应的厚度较大。此时应调节旋扭减小厚度;相反若干涉条纹有吞现象则条纹的级数在减小那么该处的等倾条纹对应的厚度较小,此时应调节旋扭增加厚度。最后直至干涉条纹稳定,无吞吐现象发生。
2.为何法珀腔的外表面与内表面不能平行?
答:若法珀腔内外表面平行的话,由内表面反射回的光线将与外表面反射光线平行,可能会发生干涉而影响实验结果。
3实验中的法珀腔介质折射率未给出是否影响实验结果?
答:由最后推出的公式可知若用1代替n会使得结果偏大。
4试说出塞满效应的应用。
汞分析仪用于测定室外、室内、生产车间以及自然界和工厂排气口的空气中的汞蒸气的浓度。加上必要的附件可以测量固、液、气各种样品。
该仪器能在静态模式和连续模式(各种交通工具)下在线监测空气中气态汞的浓度。该仪器可用来解决环境问题、石油和矿藏勘探、监测技术过程、清洁生产以及科学研
究。汞分析仪是在塞曼原子吸收光谱法的基础上应用了高频调制偏振光技术
放射源(汞灯)置于永磁体H内,汞共振线λ=254nm被分为π、σ-和σ+三种偏振塞曼组分(分别为p, s- and s+),当射线沿着磁体直向传播时,光电探测器仅仅探测s -射
线,其中一部分落在吸收线的里面,另一部分落在外界。当分析池中无气汞时,两部分s-射线的强度是一样的。当分析池中有原子吸收时,两部分s-射线的强度的差异随着汞
气浓度的增加而增加。s-射线被偏振调制器分开,s-射线的光谱移动明显小于分子吸收波段的宽度和散射范围,因此,各部分间干扰而产生的背景吸收不影响仪器的读数。10米长的多光程样品池有效的提高了仪器的灵敏度。
第二篇:西安交大高分子化学实验报告模板

 西安交通大学实验报告
西安交通大学实验报告
第 页(共 页)
课程:______________________________ 实验日期: 年 月 日
专业班号_____________组别____________ 交报告日期: 年 月 日
姓 名_____________学号____________ 报告退发: (订正、重做)
同组者_____________________________ 教师审批签字:

实验名称: 实验1 丙烯酰胺的水溶液聚合
一、实验目的
1. 了解自由基聚合的基本原理;
2. 掌握丙烯酰胺水溶液聚合的原理和方法。
二、实验原理
溶液聚合是将单体和引发剂溶于适当的溶剂中,在溶液状态下进行的聚合反应。与本体聚合相比,溶液聚合体系粘度小,传质和传热容易,聚合反应温度容易控制,不易发生自动加速现象。而且由于高分子浓度低,不易发生向高分子的链转移反应,因而支化产物少,产物分子量分布较窄;缺点是单体被稀释,单体浓度降低,聚合反应速率慢,产物分子量较低,而且如果产物不能直接以溶液形式应用,还需增加溶剂分离与回收后处理工序,加之溶液聚合的设备庞大,利用率低,成本较高。
溶液聚合在工业上常用于合成可直接以溶液形式应用的聚合物产品,如胶粘剂、涂料、油墨、浸渍剂、合成纤维的纺丝液等,而较少用于合成颗粒状或粉状产物。
聚丙烯酰胺(PAM,polyacrylamide)外观是白色固体,易吸附水分和保留水分,可以任意比例溶于水,不溶于甲醇、乙醇、丙酮、乙醚、脂肪烃和芳香烃。聚丙烯酰胺水溶液粘度随浓度的增加而急剧上升,浓度超过10%时就形成凝胶体。聚丙烯酰胺是一种水溶性高分子材料,目前广泛应用于造纸、选矿、油田开发、污水处理等,是一种优良的絮凝剂。
本实验是采用丙烯酰胺在过硫酸铵的引发下合成聚丙烯酰胺,反应方程如下:

三、仪器与试剂
1. 仪器
恒温水浴 电动搅拌器 形冷凝器 三口瓶 滴液漏斗
量筒(10mL) 烧杯(50mL、100mL)
2. 试剂
丙烯酰胺 10.0 g
过硫酸铵 0.050g
甲醇 适量
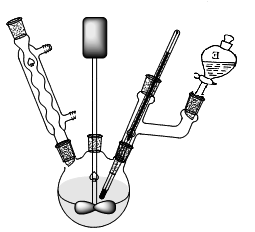 四、实验装置图及步骤
四、实验装置图及步骤
1. 按上图安装实验装置,在250mL三口瓶中加入10.0g丙烯酰胺和90mL蒸馏水,水浴加热至30℃,搅拌溶解。
2.准确称取0.050 ± 0.001g过硫酸铵,用10mL蒸馏水溶解,然后加入到三口瓶中,逐步升温到90℃,反应2~3h,冷却至室温,出料,观察所得产品的外观。并观察实验过程中的现象。
在250mL烧杯中加入60mL甲醇,在搅拌下缓缓加入上述溶液约30克(不完全沉淀处理,准确计量其中一部分!),有白色聚合物沉淀出现。静置片刻,取出沉淀,分批次沉淀。将产物置于表面皿中,在30℃下真空干燥至恒重。称重计算产率。
五、数据记录与处理
六、实验结果分析
七、思考题
1. 溶液聚合反应的溶剂应如何选择?
2. 在反应过程中,溶液的粘度是否会发生变化?为什么?
实验2 乙酸乙烯酯的乳液聚合
一、实验目的
1、掌握乳液聚合的反应特点及各组分的作用。
2、掌握乳液聚合的一般原理及实验的操作技术。
二、实验原理
本实验以乙酸乙烯酯在水介质中由聚乙烯醇(1799)和OP—10作乳化剂分散成乳液状态,用水溶性的引发剂过硫酸盐进行乳液聚合制得白乳胶。
乙酸乙烯酯(VAC)单体在过硫酸铵引发剂的作用下,按照自由基反应历程进行反应,反应式如下:

市场上的"白乳胶"就是乳液聚合方法制备的聚醋酸乙烯酯乳液。乳液聚合通常在装备回流冷凝管的搅拌反应釜中进行:加入乳化剂、引发剂水溶液和单体后,一边进行搅拌,一边加热便可制得乳液。乳液聚合温度一般控制在70~90℃之间,pH值在2~6之间。由于醋酸乙烯酯聚合反应放热较大,反应温度上升显著,一次投料法要想获得高浓度的稳定乳液比较困难,故一般采用分批加入引发剂或者单体的方法。
在乳液聚合反应中,有两种粒子成核过程,即胶束成核和均相成核。醋酸乙烯是水溶性较大的单体,28°C时在水中的溶解度为2.5%,因此,它主要以均相在核形成乳胶粒;所谓均相成核即聚合生成的短链自由基在水相中沉淀出来,沉淀粒子从水相和单体液滴上吸附了乳化剂分子而稳定,接着又扩散入单体,形成粒子。
聚合反应中可单独先用非离子乳化剂,如聚乙烯醇,OP-7,OP-10.聚乙烯醇主要起保护胶体作用,防上粒子相互并合,由于其不带电荷,对环境或介质的PH值变化不敏感,但是形成的乳胶颗粒大。单独选用阴离子型乳化剂,如烷 基磺酸钠或烷基苯磺酸钠时,由于乳胶粒外电茶的相互排斥,使乳液具有较大的稳定性,但形成的乳胶粒子小,乳液粘度大。将非离子型乳化剂和离子型乳化剂按一定比例混合使用时,常常会聚得较好的乳化效果,会使形成的乳胶粒直径比单独使用阴离子型乳化剂时的乳胶料直径大,这样就大大降低了乳胶粒表面上的电荷密度,使得带负电的离子自由基更容易进入到乳胶粒中,因而提高 了引发效率。另外,还会使两种乳化剂分子交替地吸附于乳胶粒的表面上,相当于在离子型乳化剂分子之间又锲入了非离子型乳化剂分子,这样就降低了在同一乳胶粒子上离子之间的静电斥力,增强了乳化剂分子在乳胶粒上吸附的牢度,加之非离子型乳化剂在乳胶粒子的保护作用,更使乳胶粒的稳定性得到提高。
聚醋酸乙烯乳胶广泛应用于建材,纺织,涂料等领域,主要作为胶粘剂使用.。这种用途要求其具有较好的粘接性,且粘度低,固体含量高,乳液稳定。用一般乳液聚合的一次加料方法很难做到。通常采用种子聚合方法,即分两步加料反应。第一步加入少许(如约1/3,1/5,1/10)的单体、引发剂和乳化剂进行予聚合反应,可生成颗粒很小的乳胶粒子,即种子。第二步,继续滴加单体或乳化剂单体、引发剂,在一定的搅拌条件下使其在原来形成的种子上继续长大,由此得到乳胶粒子,不仅粒度较大,而且粒度分布均匀。这样方能保证在固体含量较高的情况下,仍有较低的粘度。根据种子聚合技术,近年来具有核壳结构的高分子复合乳液有了较大发展。利用不同性能的单体制备出核、壳结构不同的聚合物,可赋予该聚合物较好的力学性质。例如:研究较多的苯乙烯-丙烯酸酯复合乳液,醋酸乙烯-丙烯酸酯复合乳液,都有很优异的性能。
醋酸乙烯的均聚物,低温下发脆,玻璃化温度高。为此,常采用外加增塑剂的方法改性,也可采用与具有柔性的单体共聚的方法,如与丙烯酸酯等共聚是有前途的发展方向。
三、实验内容
1、仪器设备:
水浴锅 电动搅拌 球形冷凝管
三口瓶 250ml 滴液漏斗 50ml
温度计0~100℃
天平 烘箱 量筒 烧杯 广泛pH试纸
2、药品及配比

四、实验步骤
(1)制备10%聚乙烯醇溶液 。称取1799加入三口瓶中 ,加热至85±1℃约半小时,液相均匀透明,降温至65℃备用。0.5 g(NH4)2S2O4溶于10 ml 水中。
(2)内有聚乙烯醇溶液的三口瓶中加入定量水及OP—10搅拌20min,温度控制在66~68℃。
(3)加入10g VAC占总量40%的引发剂(0.2g)。搅拌10min,升温到70℃,控制回流。当回流消失后升温至80℃。滴加30g VAC。视回流快慢,控制滴加速度约0.5~1小时滴完,并在此期间把余下引发剂的一半加入,单体滴加完后,加入最后的引发剂,再搅拌5min。然后滴加最后10g VAC。
(4)投料完毕,继续加热回流,缓慢升温以不产生大量泡沫为准,最后升温至90℃,反应至无单体回流为止,反应过程中,可加入少量碳酸氢钠溶液调PH值为4~6。冷却到50℃。加入增塑剂邻苯二甲酸二丁酯(DBP)搅拌10min出料。
(5)测含固量。取2g乳液(0.0002g)置于至恒重的玻璃表面皿中,放于110℃烘箱中至恒重计算含固量(约4小时)。
含固率=干燥后样品重/干燥前样品重×100%
(6)计算产率:反应完毕后,将一部分胶乳(20ml)加入50ml饱和氯化钠溶液破乳,把聚合物沉淀出来,干燥,粗略计算产率。若加入50ml饱和氯化钠溶液后还不能破乳,则需要再加入更高浓度的氯化钠溶液(将水加热至60℃左右后溶解氯化钠制成60℃下的饱和氯化钠溶液)
转化率(%)=(Wc-SWb/Wa)/(GWb/Wa)
式中:Wc 取样干固重量
S 实验中加入的乳化剂、引发剂、增塑剂总重量
Wa 三口瓶内乳液体系总重量
Wb 取样重量(干燥前)
G 实验中醋酸乙烯单体加入总重量
五、数据记录与处理
六、实验结果分析
七、思考题
1、什么叫乳液聚合,有何特点?
2、为什么要严格控制单体滴加速度和聚合反应温度?
3、比较乳液聚合与本体聚合有什么区别?
-
塞曼效应实验报告完整版
南昌大学物理实验报告学生姓名学号5502210039专业班级应物101班实验时间教师编号T017成绩塞曼效应一实验目的1观察塞曼效…
-
塞曼效应实验报告
近代物理实验报告塞曼效应学院班级姓名学号时间塞曼效应摘要本实验利用高分辨光谱仪器法布里珀罗FabryPerot标准具研究汞5461…
-
近代物理实验 塞曼效应
深圳大学实验报告课程名称:大学近代物理实验实验名称:塞曼效应实验地点:实验时间:实验报告提交时间:一、实验目的1.提高物理想象能力…
-
西安交大《塞曼效应实验报告》
塞曼效应1896年荷兰物理学家塞曼PZeeman在实验中发现当光源放在足够强的磁场中时原来的一条光谱线会分裂成几条光谱线分裂的条数…
-
塞曼效应实验报告
实验题目塞曼效应实验目的研究塞曼分裂谱的特征学习应用塞曼效应测量电子的荷质比和研究原子能级结构的方法实验仪器塞曼效应实验平台仪器磁…
-
计算机仿真物理实验1—塞曼效应实验报告模板
计算机仿真物理实验1塞曼效应实验报告模板班实验组号姓名学号课程名称大学物理实验220xx年9月3日实验题目计算机仿真物理实验1塞曼…
-
近代物理实验报告—塞曼效应
塞曼效应摘要本实验主要运用光栅摄谱仪拍摄Hg在磁场中与无磁场中的谱线了解Hg谱线的分裂情况并利用Fe做比较光谱用阿贝比长仪测量并计…
- 塞曼效应_预习报告
-
塞曼效应与法拉第效应实验报告
塞曼效应与法拉第效应实验报告摘要法布里珀罗标准具的工作原理与调节使用方法通过法布里珀罗标准具观测汞原子5461nm谱线的分裂现象以…
-
塞曼效应报告
西安交通大学实验报告第页共页课程近代物理实验实验日期年月日专业班号组别交报告日期年月日姓名学号报告退发订正重做同组者教师审批签字实…