医疗器械风险管理报告格式
医疗器械风险管理报告格式一(正相关法)
1前言
本文是对XXXX进行风险管理的报告,报告中对所有的可能危害以及每一个危害产生的原因进行了判定。对于每种危害可能产生损害的严重度和危害的发生概率进行了估计。在某一风险水平不可接受时,采取了降低见的控制措施,同时,对采取风险措施后的剩余风险进行了评价。最后,使所有的剩余风险的水平达到可以接受。
2 适用范围
本报告适用于XXXX产品,该产品处于设计和开发阶段(或处于小批生产阶段)。它由X个部分组成:(具体说明)
其中,有一些外购、外协件(具体说明)
3应用资料
3.1相关标准
1) YY0316-2003医疗器械——风险管理对医疗器械的应用
2) GB9706.1-1995医用电气设备 第一部分:通用安全要求;
3) IEC60601-1-4:1996医用电器设备——第一部分:通用安全要求——4:并行标准:医用可编程电气系统
4) 产品标准及其他
3.2 有关产品的资料
1)使用说明书
2)医院使用情况、维修记录、顾客投诉、意外事故记录等
3)专业文献中的文章和其他信息
3.3缩写字
1) ALARP区:合理可靠降低区
2) FMEA:失效模式和效应分析
3) FMECA:失效模式、效应和危害度分析;
4) FTA:故障树分析等;
5) SEEA:software errors effect analysis
4 产品说明
4.1概况
本风险管理的对象是。。。。。。(如能加入照片或图片最好)
4.2功能说明
如果后面的风险降低控制措施需要对某些功能有所了解,应对相关功能做出说明。
4.3 预期用途
4.4使用环境
4.5风险评估相关数据及资料
5 风险管理的产品说明
5.1概况
本风险管理的对象是。。。。。。(如能加入照片或图片最好)
5.2功能说明
如果后面的见险降低控制措施需要对某些功能有所了解,应对相关功能做出说明。
5.3 预期用途
5.4 使用环境
5.5 风险评估相关数据及资料
6 风险管理过程的实施
6.1 第1步:预期目的和定性、定量特性的判定
6.2 第2步:已知或预见危害的判定
6.3 第3步:风险估计
6.3.1 损害的严重度准则和严重度估计
6.3.2 每项损害的原因判定
先利用专业知识直观地寻找潜在原因,进一步的原因分析则可应用FMEA、FTA方法。
6.3.3发生概率准则和估计
6.3.4风险估计
6.4第四步:风险评价
在采用ISO/IEC JWG——RMN10,ISO/TC210——IEC/SC62A联合工作组1997.02的格式时:
表1 风险评价表

6.5 第五、六步:采取风险控制措施
6.6 第七步:剩余风险评价
6.7第八步:风险/受益分析
6.8 第九、十、十一步:
6.9 第十二步:风险管理报告
附录
表2 采取控制措施以前风险水平

表3 采取控制措施以后风险水平

6.10 控制措施总结
6.11控制措施的验证和评审
第二篇:风险管理报告
247多参数诊断仪
风险管理报告
(模版)
编 写: 魏广道
风险管理参加人员:魏广道/李湘辉/张丕治
日 期: 20## 年 3 月 20 日
评 审: 李湘辉/张丕治
日 期: 20## 年 3 月 21 日
批 准: 张丕治
日 期: 20## 年 3 月 29 日
顺泰医疗器材(深圳)有限公司(盖章)
目 录
第一章 概述
第二章 风险管理人员及其职责分工
第三章 风险可接受准则
第四章 预期用途和与安全性有关的特征的判定
第五章 判定可预见的危害、危害分析及初始风险控制方案
第六章 风险评价、风险控制和风险控制措施验证
第七章 综合剩余风险评价
第八章 生产和生产后信息
第九章 风险管理评审结论
第一章 概述
1. 编制依据
1.1 相关标准(按企业所生产产品的类型列举相关标准,以下标准为举例)
1) YY0316-2008医疗器械——风险管理对医疗器械的应用
2) GB/T9706.1-2007医用电气设备 第一部分:通用安全要求
3) GB9969.1 工业产品使用说明书 总则
4) GB/T14710-2009 医用电器设备环境要求及试验方法
5) YY0670-2008 无创自动测量血压计
6) YY0784-2010 医用电气设备_医用脉搏血氧仪设备基本安全和主要性能专用 要求
7) 注册产品标准(247多参数诊断仪YZB/粤-2013)
8) YY0466.2009 医疗器械 用于医疗器械标签、标记和提供信息的符号
9) 其他标准
1.2 产品的有关资料
1) 247多参数诊断仪说明书
2) 公司其他类似产品医院使用情况、维修记录、顾客投诉、意外事故记录等
3) 专业文献中的文章和其他信息
2. 目的和适用范围
本文是对247多参数诊断仪进行风险管理的报告,报告中对247多参数诊断仪在上市后风险管理情况进行总体评价,所有的可能危害以及每一个危害产生的原因进行了判定。对于每种危害可能产生损害的严重度和危害的发生概率进行了估计。在某一风险水平不可接受时,采取了降低见的控制措施,同时,对采取风险措施后的剩余风险进行了可接受性评价,证实对产品的风险已进行了管理,并且控制在可接受范围内。
4) 本报告适用于247多参数诊断仪
产品,该产品处于批量生产阶段。
3. 产品描述
本风险管理的对象是 多参数血压诊断仪,包括 247 、247#1、247#2 、247#3

适应症:该诊断仪是一种临床级血压、心率、平均动脉压、血氧和体温的无创诊断仪。适合在医院、医疗机构、门诊、诊所以及其他亚急性环境中使用。
禁忌症:该产品只用于成人和儿童。请勿将本设备用于 3 岁以下儿童、婴儿或新生儿患者。
设备由以下部分组成:
247 诊断仪由血压模块、体温模块、血氧模块、AC/DC电源适配器、血压袖带、血氧探头 和一次性探针帽、充电电池(可选项)组成。
247#1 诊断仪由血压模块、AC/DC电源适配器、血压袖带、充电电池(可选项)组成。
247#2诊断仪由血压模块、体温模块、AC/DC电源适配器、血压袖带和一次性探针帽、充电 电池(可选项)组成。
247#3诊断仪由血压模块、血氧模块、AC/DC电源适配器、血压袖带、血氧探头和充电电池(可选项)组成。
风险管理计划及实施情况简述
247多参数诊断仪产品于20##年开始策划立项。立项同时,我们就针对该产品进行了风险管理活动的策划,指定了风险管理计划(文件编号:XXXX,版本号XX)。
该风险管理计划确定了风险管理活动范围、参加人员及职责和权限的分配、基于制造商决定可接受风险方针的风险可接受性准则,包括在损害发生概率不能估计时的可接受风险的准则、风险管理活动计划等内容。
247多参数诊断仪产品于20##年开始批量生产,未发生设计、材料、工艺等方面的变更
第二章 风险管理人员及其职责分工
风险管理小组(team):

第三章 风险可接受准则
1.风险的严重度水平

2.风险的概率分级
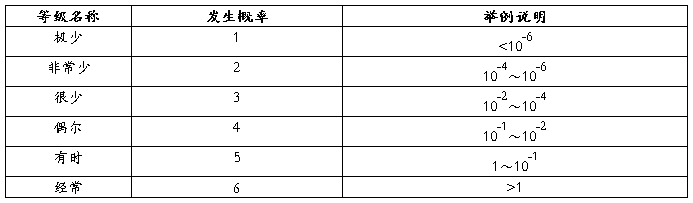
3.风险评价准则
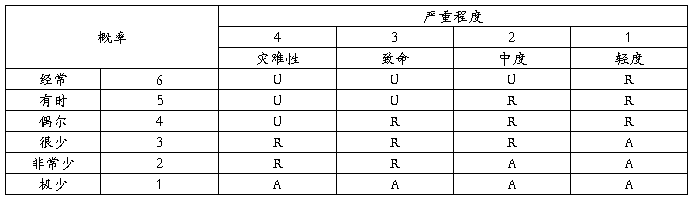
说明:A:可接受的风险;R:合理可行降低(ALARP)的风险;U:不经过风险/收益分析即判定为不可接受的风险
第四章 预期用途和与安全性有关的特征的判定
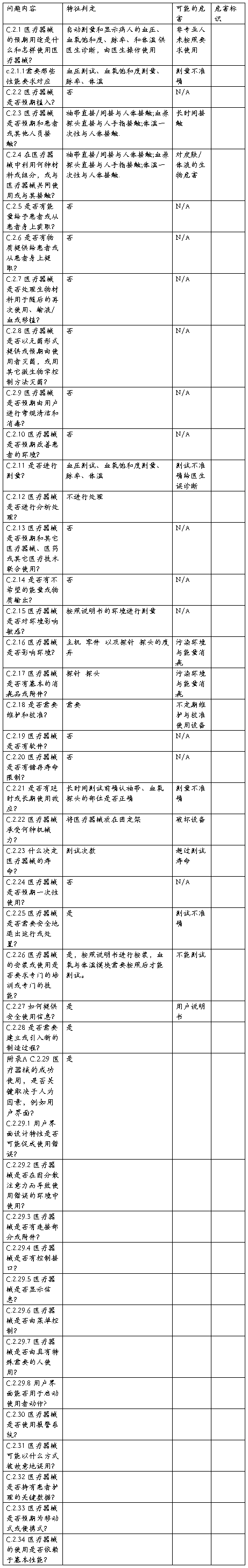
企业以YY0316-2008附录C为基础对医疗器械预期用途和与安全性有关的特征进行了判定,通过对涉及医疗器械的制造、预期使用者、预期用途、合理可预见的误用和最终处置等等提出一系列问题的方法,逐步了解该产品的安全性特征,为进一步的风险分析打下基础,XXXX产品安全特征问题清单如下:
表 1 多参数诊断仪器产品安全特征问题清单
第五章 判定可预见的危害、危害分析及初始风险控制方案
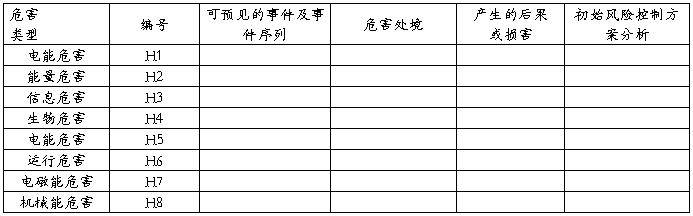
企业在对危害分析中,已考虑合理可预见的情况,它们包括正常条件下、故障条件下;对危害产生的后果或损害包括:对于患者的危害、对于操作者的危害、对于维修人员的危害、对于附近人员的危害、对于环境的危害。XXXX产品的初始危害分析表见表2,包括可预见的事件序列、危害处境和可发生的损害及初始风险控制方案分析
表2:XXXX产品的初始危害分析表
。。。。。。
第六章 风险评价、风险控制和风险控制措施验证
公司对已知危害进行风险评价,按照风险可接受准则判断每个危害的风险是否达到可接受水平,对合理可行降低的风险、不经过风险/收益分析既判定为不可接受的风险采取控制措施,并对具体措施进行实施验证,同时重新对采取措施后的风险进行估计,确认其风险水平是否可接受。XXXX产品风险评价、风险控制措施记录表见表3:
表3 XXXX产品风险评价、风险控制措施记录表见表
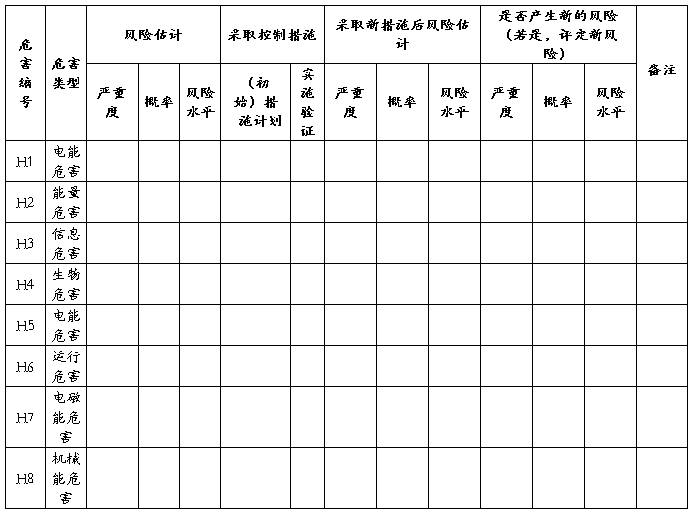
。。。。。。
第七章 综合剩余风险评价
公司在采取降低风险的措施后,……等危害的风险已降到广泛可接受的程度,……等危害的风险也降到了合理可行降低的程度。(还要说明采取降低风险的措施后,有没有引入新的风险,若有,则须进行再次评价和控制),经评审小组确认:产品综合剩余风险可接受。具体评价方面:
1)单个风险的风险控制是否有相互矛盾的要求?
结论:尚未发现现有风险控制有相互矛盾的情况。
2)警告的评审(包括警告是否过多?)
结论:警告的提示清晰,符合规范。
3)说明书的评审(包括是否有矛盾的地方,是否难以遵守)
结论:产品说明书符合10 号令及产品专用安全标准要求,相关产品安全方面的描述清晰易懂,易于使用者阅读。
4)和同类产品进行比较
结论:通过与XXX 公司的XXX 型XX产品进行的临床、性能、功能比较比较认为产品与目前市场上反映较好的XXX 公司的XXX 型XX产品从性能指标到功能及临床使用上是相同的。
5)与类似功能产品进行比较(必要时)
6)专家结论
结论:风险管理评审小组在分析了以上方面,并临床应用专家进行了充分的沟通后,一致评价,本产品的综合剩余风险可接受。
第八章 生产和生产后信息
医疗器械XXXX产品已正式投入生产并上市,公司已对生产和生产后信息收集和评审,并填写了XXXX产品生产和生产后信息收集表(表4),以决定是否需要改进产品(尤其安全性)和服务
该项目风险管理负责人对得到的生产和生产后信息进行管理,必要时,风险管理小组开展活动实施动态风险管理。
表4:XXXX产品生产和生产后信息收集表
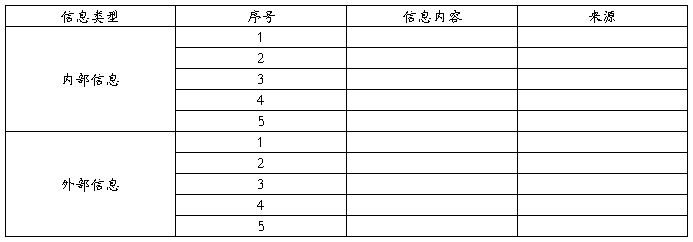
第九章 风险管理评审结论
风险管理评审小组经过对XXXX产品评审,认为:
- 风险管理计划已被适当地实施;
- 综合剩余风险是可接受的;
- 已有适当方法获得相关生产和生产后信息,并在适当时启动动态风险管理程序。
XXXX产品全部剩余风险处于风险可接受准则的可接受范围内,且收益超过风险。
签名:
-
医疗器械风险分析报告
安全风险分析报告依据YY031620xx医疗器械风险管理对医疗器械的应用单位名称医疗器械有限公司产品名称医用纱棉块申报日期20xx…
-
医疗器械风险管理报告格式
医疗器械风险管理报告格式一正相关法1前言本文是对XXXX进行风险管理的报告报告中对所有的可能危害以及每一个危害产生的原因进行了判定…
-
医疗器械风险管理报告(注册版)
医疗器械风险管理报告XXXXX调整仪批准批准日期Xxxxx医疗器械科技有限公司目录第一章综述1第二章风险管理评审输入2第三章风险管…
-
医疗器械风险管理报告
医疗器械风险管理报告有源医疗器械医疗器械风险管理报告XXX型XX诊断仪文件编号批准人批准日期AAAA医疗器械有限公司1医疗器械风险…
-
医疗器械产品风险分析报告范例
无线心电和体温监测仪产品风险分析报告中科康馨电子技术北京有限公司20xx年11月理疗仪产品风险分析报告1一产品预期用途预期目的和与…
- 医疗器械风险管理计划
-
医疗器械产品风险分析报告范例
产品风险分析报告广州市XX医疗器械有限公司20XX年6月一、产品预期用途/预期目的和与安全性有关的特征的判定按照《YY/T0316…
-
医疗器械产品风险分析报告范例
无线心电和体温监测仪产品风险分析报告中科康馨电子技术北京有限公司20xx年11月理疗仪产品风险分析报告1一产品预期用途预期目的和与…
-
医疗器械风险管理报告(20xx)
风险管理报告编制人批准人批准日期目录第一章综述1第二章风险管理评审输入5第三章风险管理评审7第四章风险管理评审结论8附件19附件2…
-
医疗器械产品--安全风险分析报告
血清碱性磷酸酶测定试剂盒ALP安全风险分析报告1总则血清碱性磷酸酶测定试剂盒以下简称ALP测试盒是一种临床检验体外诊断化学试剂中酶…
-
风险管理报告模板
风险管理报告产品基本信息编制人批准人批准日期目录第一章综述1第二章风险管理评审输入5第三章风险管理评审7第四章风险管理评审结论8附…