出生缺陷诊断制度
出生缺陷诊断制度
出生缺陷是指胚胎或胎儿发育过程中结构或功能发生的异常,出生缺陷影响了我过人口素质,给家庭和社会带来沉重负担,已引起政府和社会各界的高度重视,为初步摸清全国出生缺陷的发生状况及其变化趋势,故出生缺陷诊断尤为重要,因此特制定如下诊断制度
1、 成立出生缺陷监测专家组,负责辖区内出生缺陷病例的确认及技术指导
2、 出生缺陷应由县级及以上医疗机构诊断,并经专家组确认部分体表畸形如多指、脐疝
等可由乡卫生院诊断。
3、 产前诊断的出生缺陷必须在出生后进行确认但由具有产前诊断资质的医疗机构在产
前确诊的致死性,重大出生缺陷和染色体异常应计为真病例
第二篇:出生缺陷诊断制度
出生缺陷诊断制度
为提高出生缺陷的诊断水平,避免误诊、漏诊,根据《中国出生缺陷监测方案》、《辽宁省出生缺陷医院监测方案》和《辽宁省出生缺陷诊断制度》制定我区出生缺陷诊断制度:
一、各乡镇卫生院负责本辖区内出生缺陷监测工作的监督和管理;协调辖区内各级医疗保健机构实施出生缺陷诊断制度。
二、设县区卫生行政部门应成立出生缺陷诊断专家组,成员由产科、计划生育科、儿科、内分泌科、外科、物理诊断科(如超声科、放射科)、病理科等组成,负责全区及辖区内疑难出生缺陷病例的诊断。
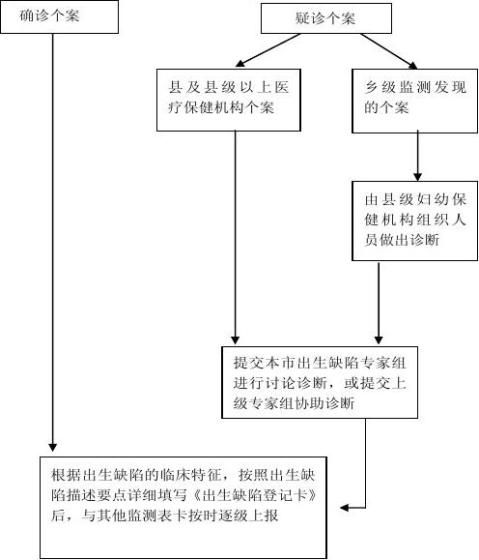
三、各级妇幼保健机构和出生缺陷的监测医院发现出生缺陷应根据出生缺陷的临床特征(见附件1)进行诊断,对体表可见的或非体表可见的出生缺陷,应根据出生缺陷的描述要点(见附件2)详细描述记录并填写《出生缺陷登记卡》上报,对不能确诊的病例应提交本区出生缺陷专家组进行讨论诊断,或提交上级出生缺陷诊断专家组协助诊断。(见附件3)
四、人群监测点的乡级监测人员发现并诊断的出生缺陷,应填写《出生缺陷儿登记卡》上报,对不能明确诊断的出生缺陷应上报县级妇幼保健机构,由县级妇幼保健机构组织人员根据收集的资料或入户调查,做出诊断。对不能确诊的病例应提交上级出生缺陷诊断专家组协助诊断。
五、经超声检查发现的可疑胎儿异常或实施治疗性引产的出生缺陷应由具备产前诊断资质的医疗保健机构进行产前诊断。 1
附件1:出生缺陷的诊断 附件2:出生缺陷描述要点 附件3:出生缺陷诊断流程 2
附件1.出生缺陷的诊断
主要出生缺陷的临床特征
1. 无脑畸形
无脑畸形(Anencephaly)是以颅骨穹隆及其覆盖的皮肤和脑的全部或部分缺如为特征的先天性畸形。应排除无头畸形和积水性无脑。无脑畸形患儿主要表现为颅骨穹隆(包括枕骨、顶骨、额骨)缺如,覆盖颅骨的皮肤缺如或存在;大脑半球或额叶缺如,脑干和小脑裸露;头顶平坦,颅面比例失调,由正常的2:1变为1:2或1:3;患儿眼珠突出,眼距宽,低位耳,呈“蛙样”面容。无脑畸形的缺损可以前至眉弓,后至枕骨大孔,称全无脑畸形;但也可以范围较小,只在头顶部出现一个较小的洞,称部分无脑畸形;有时,前至头颅,后至脊柱部分或全部均未闭合,称为颅脊柱裂。一般为死胎/死产或出生后不久即死亡,不可能存活。产前自妊娠16周即可根据B超和孕母血清或羊水甲胎蛋白测定做出诊断。出生后通过肉眼观察即可做出诊断。
2. 脊柱裂
脊柱裂(Spina bifida)以脊膜和/或脊髓通过未完全闭合的脊椎疝出或暴露于外为特征的先天性畸形。应排除隐性脊柱裂及无神经管闭合不全的骶尾部畸胎瘤。临床上根据畸形发生部位分为颈段、胸段、腰段和骶段脊柱裂。根据膨出物的性质可分为:①脊膜膨出;②脊膜脊髓膨出;③脊髓外翻。前二者在畸形发生部位可见一囊状物,囊状物大小不等,质软或软中有实物,覆盖的皮肤趋向变薄、变软,表面有液珠渗出。脊髓外翻表现为局部有两平行的红色肉芽面。产前根据
B超和孕母血清或羊水甲胎蛋白测定即可诊断。出生后通过肉眼观察即可做出诊断。
3. 脑膨出
脑膨出(Encephalocele)是以脑膜/脑通过颅骨裂膨出为特征的先天性畸形。脑膨出常发生在枕部,也可见于鼻根部、额部、顶部、颞部等。根据膨出内容可分为:①脑膜膨出;②脑膜脑膨出。临床可见脑部膨出的囊性肿物大小不等,可扪及矢状缝增宽,患儿啼哭时肿物可增大甚至可扪及血管搏动。脑膜膨出在暗室用手电筒局部透射时呈一片红色,局部穿刺可抽出正常脑脊液。产前B超检查有助诊断。出生后通过肉眼观察即可做出诊断。
4. 先天性脑积水
先天性脑积水(Congenital hydrocephalus)是以脑室系统扩大伴以脑脊液梗阻为特征的先天性畸形,不伴有原发性脑萎缩,伴有或不伴有头颅增大。患儿可表现为头颅增大,尤以额部为甚,颅面比例大于2:1;头皮变薄、发亮,静脉怒张;前囟宽大、突出、搏动消失,后囟及侧囟扩大,颅骨缝裂开,叩诊前囟四周呈破罐声;双眼珠向下,白色巩膜显露(落日征)。产前B超检查可发现颅骨径增大,侧脑室扩大。出生后通过肉眼观察可做出初步诊断,脑部X线摄影、B超、CT检查可进一步确诊。
5. 腭裂
腭裂(Cleft palate)是以切牙孔后的硬腭和软腭处存在裂隙为特征的先天性畸形,包括黏膜下腭裂,即隐性腭裂。注意排除功能性
短腭和高而窄的腭。临床上根据其分裂程度可分为3度:Ⅰ度,腭垂裂或软腭裂;Ⅱ度,软腭全裂,硬腭部分裂开,但没有达牙槽骨(牙槽突);Ⅲ度,软腭、硬腭全裂,直达牙槽骨。出生后通过肉眼观察即可做出诊断。
6. 唇裂
唇裂(Cleft lip)是以上唇线在正中线外侧裂开为特征的先天性畸形。临床上分为单侧和双侧,以单侧多见。根据唇裂程度可分为3度:Ⅰ度,红唇裂;Ⅱ度,红唇裂,皮肤部分裂,没达鼻底;Ⅲ度,红唇裂,皮肤全裂,直达鼻底。出生后通过肉眼观察即可做出诊断。
7. 唇裂合并腭裂
唇裂合并腭裂(Cleft lip with cleft palate)是以上唇裂伴有牙槽嵴裂和腭裂为特征的先天性畸形。出生后通过肉眼观察即可做出诊断。
8.先天性耳廓畸形
先天性耳廓畸形(Congenital malformation of auricle)是以耳廓的大小、形态及位臵异常为特征的先天性畸形。临床主要表现为不同程度的耳廓过度发育或发育不良,如巨耳、小耳、副耳、颊耳、并耳等。该畸形可单独发生,但常伴外耳道、中耳、内耳畸形。出生后通过肉眼观察即可做出诊断。
9.食管闭锁或狭窄
食管闭锁或狭窄(Congenital esophageal atresia)是以食管闭锁或狭窄合并或者不合并食管气管瘘为特征的先天性畸形,包括单纯
食管气管瘘。患儿出生后以反复发作进食后呛咳、发绀,并伴有唾液过多为特点。严重者可致肺炎、呼吸困难,甚至呼吸功能衰竭。吸出分泌物后,发绀缓解。但过一段时间后,分泌物可再次堵塞气管导致发绀发生,如此反复。先天性食管闭锁者,唾液过多,可从口鼻溢出,而且发绀的发生往往较单纯食管气管瘘者频繁、严重。根据典型的临床症状即可做出诊断。鼻饲管插入受阻,食管碘油造影胸腹部X线检查可进一步明确诊断。
10. 直肠肛门闭锁或狭窄
直肠肛门闭锁或狭窄(Congenital atresia of rectum and anus)是以肛门缺如、直肠闭锁(狭窄)或直肠肛门闭锁(狭窄),与邻近器官有瘘或无瘘为特征的先天性畸形。临床表现为低位肠梗阻,主要分为以下5种类型:
(1)肛门直肠低位闭锁:闭锁的直肠盲端距会阴不超过2cm厘米,会阴中央肛门部位有完整的皮肤,会阴发育不良,中央有凹陷,婴儿啼哭隆起,扪及冲击感。
(2)肛门膜状闭锁:肛门区有薄膜覆盖并向外膨出。
(3)肛门狭窄或肛管直肠交界处狭窄:大便呈细条状或表现为不完全性肠梗阻。
(4)肛门直肠高位闭锁:闭锁的直肠盲端距会阴皮肤在2cm以上,会阴中央肛门区有完整的皮肤覆盖。
(5)直肠闭锁或狭窄:肛门外表正常,但直肠下端闭锁或狭窄,无胎粪或少量,肛门指检受阻。
以上各型可与泌尿生殖器官相通形成瘘管,男婴有瘘管者可达75%,女婴可达90%。男婴瘘管常见:直肠膀胱瘘、直肠尿道瘘、直肠会阴瘘。女婴瘘管常见:直肠阴道瘘、直肠前庭瘘、直肠会阴瘘。
11.尿道下裂
尿道下裂(Hypospadias)是以尿道异常开口于阴茎、阴囊腹侧或会阴为特征的先天性畸形。应排除尿道上裂、阴茎弯曲或包皮茎过长伴有正常尿道开口以及假两性畸形。临床根据尿道开口的位臵可分为:①阴茎头型;②阴茎体型;③阴茎阴囊型;④会阴型。出生后通过肉眼观察即可做出诊断。
12.膀胱外翻
膀胱外翻(Extrophy of urinary bladder)是以下腹壁部分缺损、膀胱前壁裂开、后壁暴露于外为特征的先天性畸形。患儿表现为前腹壁及耻骨联合没有融合,形成一裂隙;膀胱黏膜外翻呈鲜红色,其下方可见输尿管开口,有尿液自此外溢。出生后通过肉眼观察即可做出诊断。
13.先天性马蹄内翻足
先天性马蹄内翻足(Congenital talipes equinovarus)是以全足内翻、前足内收、跗骨间关节跖屈为特征的先天性畸形。其典型表现为①足内侧缘向内上方翻转;②前足内收;③距小腿关节(踝关节)和跗骨间关节跖屈。出生后根据临床表现即可做出初步诊断,进一步做足部X线摄影检查可确诊。
14. 多指(趾)
多指(趾)(Polydactyly)是以一手(足)或双手(足)多发生一个或多个手(足)指(趾)样物为特征的先天性畸形。应排除指甲裂开、指端呈分叉状病变。临床上可表现为:①仅赘生一圆球形的软组织,无骨骼、肌及关节,与手指(足趾)间的连接为一凹陷的环形束状带;②赘生的手指(趾)外形较完整,但常只有两节指(趾)骨,可与掌骨间有关节,无活动功能;③多余手指(趾)近乎正常,有三节指骨和一个发育差的掌骨,较少见。出生后通过肉眼观察即可做出诊断。
15.并指(趾)
并指(趾)(Syndactyly)是以指(趾)与指(趾)间的皮肤、皮下软组织或骨组织相连为特征的先天性畸形。其临床表现如下:①相邻二指(趾)间皮肤、皮下软组织甚至骨组织相连,多发生在中指和环指;②示指至小指连成一块;③交叉性并指(趾),即非邻近指(趾)与指(趾)间发生融合。根据皮肤与骨骼的连接情况,可分为两类:①软组织并指(趾);②骨骼并指(趾)。出生后通过肉眼观察即可做出诊断。
16.肢体短缩
肢体短缩(Limb reduction defects)是以一个或几个肢体骨骼完全缺如或部分缺如或严重发育不良为特征的先天性畸形。根据其临床表现可分为以下几种类型:
(1)横向短缩:又称先天性截肢,肢体部分或全部缺如,常为单侧性。
(2)纵向短缩:肢体的桡骨或尺骨缺如或严重发育不良,或手、足中央纵裂。
(3)中段缺如:又称“海豹畸形”,肢体远端基本正常,近端缺如或严重发育不良,常为双侧性。
(4)多发性肢体短缩畸形:同时有上述几种短缩畸形。
产前B超可诊断严重的肢体短缩畸形,出生后通过肉眼观察或X线摄影即可诊断。
缺指(趾)(Ectrodactyly)是以一个或多个手指(趾)部分或完全缺如为特征的先天性畸形,属肢体短缩畸形一类。常见以下3种表现:①一个或多个手指(趾)部分或完全缺如;②桡侧纵裂缺如;③中线纵裂缺如,形成裂手畸形。
裂手(足)(Split hand,Split foot)是以手(足)中央纵列部分发育不良及缺如为特征的先天性畸形,属肢体短缩畸形一类。裂手(足)多见于双侧,常伴有不同程度的并指(趾)和缺指(趾)。典型的裂手以中指的缺如最为多见,可以是部分缺如,也可以是包括掌骨在内的全部缺如。剩余的桡、尺侧的手指分别相互融合而使手纵裂为桡、尺两部分。出生后通过肉眼观察即可做出诊断。
17.先天性膈疝
先天性膈疝(Congenital diaphragmatic hernia)是以膈肌发育不良,腹腔器官从膈肌缺损或薄弱部位进入胸腔,胸膜作为疝囊为特征的先天性畸形。根据膈疝的发生部位,主要分为两种类型:①胸腹裂孔疝;②胸骨后疝。其主要临床表现有:①呼吸困难,并可伴呕吐;
②舟状腹,患侧呈桶状胸,纵隔移位,患侧胸部呼吸音降低,可听到肠鸣,叩浊。根据临床表现可做出初步诊断。胸部X线检查或用水溶性造影剂做胃肠造影,或B型超声检查,尸体解剖等均有助于明确诊断。
18. 脐膨出
脐膨出(Omphalocele)是以脐根部腹壁缺损, 腹腔器官从缺损处疝出为特征的先天性畸形。应排除腹裂、腹壁肌发育不全和脐疝等。根据缺损环的大小,可分为巨型(直径大于5cm)和小型(直径小于5cm)脐膨出。患儿表现为腹腔器官从脐部周围缺损处疝出,由腹膜和羊膜构成的囊膜完整或不完整地覆盖于上。通过此半透明的囊膜可以见到所疝出的器官(多为小肠,结肠及肝),故称“玻璃腹”。有时囊膜可发生破裂而使器官从破裂处脱出。出生后通过肉眼观察即可做出诊断。
19.腹裂
腹裂(Gastroschisis)又称脐旁裂,是以腹腔器官通过腹壁缺损处暴露于外为特征的先天性畸形。应排除脐膨出。患儿表现为部分腹腔器官暴露于外,无疝囊覆盖。暴露肠管常有水肿、增厚。色泽发紫,肠襻相互粘连,没有肠蠕动,少数病例肠管已坏死。裂口以位于脐部右侧多见。脐部和脐带正常。按发生部位可分:①上腹裂,主要内容物为肠管、胃和肝;②下腹裂,常见膀胱外翻;③中腹裂和全腹裂。产前B超检查和孕母血清或羊水甲胎蛋白(AFP)测定有助于诊断。出生后通过肉眼观察即可做出诊断。
20. 联体双胎
联体双胎(Conjoined twins)指单卵孪生胎儿间的某一部位不能分离而互相连接的一种先天性畸形。联体双胎的临床表现多种多样。根据其连接形态,可分对称性和非对称性连接;依其连接部位不同,可有腹侧相连、背侧相连、头部相连和尾部相连等。常见以下几种类型:①胸部联体,最常见,为胸骨或胸骨附近相连接;②脐部联体,较常见,两畸形儿面对面,常发生脐,前腹壁,肝胆系统,上、下消化道以及泌尿系统等组织器官共有;③臀部联体,两畸形儿背靠背相连。④坐骨联体,较少见,两畸形儿脊柱轴相对在一条直线上,有时可见下肢仅3条腿,共用1个肛门。⑤头部联体,其连接部位在头顶部、枕部或顶骨旁;⑥寄生胎,一个个体发育完整、大小正常,而另一个则仅为部分胎儿,常相连在正常儿的腹部,也可寄生在背部、骶部或体腔内等。出生后通过肉眼观察即可做出诊断。
21.唐氏综合征
唐氏综合征(Down综合征)又称21-三体综合征(Trisomy 21 syndrome)、先天愚型。患者第21对染色体比正常人多出一条而导致的以智力发育障碍为主要特征的综合征。其主要特征如下:①眼距宽(>2.5cm);②塌鼻;③外眦上斜;④全身肌张力低;⑤双侧或单侧通贯手和/或高位三叉点t;⑥小指短小,其中节短或缺如;⑦Ⅰ,Ⅱ趾距宽(草鞋足);⑧拇趾球部胫侧弓状纹;⑨低位耳。染色体分析:以核型47,XX(或XY)+21最常见,占94%~95%;1%~2%为21-三体嵌合型;2%~3%为易位型,包括D/G易位和G/G易位。以上特征中有5
项组合的病例再结合其他特点可初步筛查为唐氏综合征,确诊需作染色体检查。
22. 先天性心脏病
先天性心脏病(Congenital heart defects)是一大类疾病的总称,包括室间隔缺损、房间隔缺损、大血管异位、法洛四联症等。
(1)动脉导管未闭(Patent ductus arterious):是指婴儿在出生后4个星期左右,动脉导管未闭合而形成主动脉与肺动脉之间先天性异常的通道。导管细小者症状常不明显,多在查体时发现。大导管者因大量血液“自左向右分流”,在婴儿期即可因左心室衰竭而产生急性呼吸困难、呼吸道感染、体重不增。动脉导管未闭并发肺动脉高压和逆向分流的病例,则出现劳累后气急、发绀等症状。本症的临床表现较为典型,约95%的病例根据典型的临床表现即可做出诊断,但有少数可疑病例,需要做心导管检查和逆行主动脉造影方能明确诊断。
(2)房间隔缺损(Atrial septal defect):是原始心房分隔过程的异常,在左右心房间仍残留未闭之房间孔。其中第二房间孔(继发孔)型房间隔缺损是最常见的先天性心脏畸形之一。大部分患儿早期无症状,出现病状时,常见的是活动后心悸、气短和乏力。在成人患者出现气短主要是由于肺动脉高压和心力衰竭所至。房性心律失常通常在40岁左右出现,可能是由于右心房肥厚或心力衰竭所致。临床上根据症状、体征及心电图检查结果可做出初步诊断。结合胸部X线摄影及超声心动图检查结果可明确诊断。
(3)室间隔缺损(Ventricular septal defect):是由于胚胎发
育不全造成心室间隔部位的异常交通,并在心室水平出现左向右分流的先天性心血管畸形。该畸形可以单独存在,也可同时合并其他畸形。室间隔缺损有自然闭合的趋势。室间隔缺损患儿生后1年内缺损可完全自然闭合或有所缩小,这一现象可以解释为何巨大室间隔缺损在成人并不常见。巨大室间隔缺损由于高压性肺血管疾病易致肺血管阻力增高,随患者年龄增加而逐渐加重。本病根据病史、体征,以及X线、心电图和超声心动图检查结果诊断不甚困难,再结合心导管和心血管造影检查结果可确定诊断。
(4)房室间隔缺损(Atrio ventricular septal defect):又称心内膜垫缺损房室管畸形和共同房室通道,现统称为房室间隔缺损。其主要特点是房室瓣上下间隔发育不全或缺如,以及房室瓣发育畸形。单纯型房室间隔缺损临床症状与继发性房间隔缺损相同,有运动性心悸、气短,易发生呼吸道感染。而部分和完全性房室间隔缺损症状出现较早,病情进展快,心脏明显扩大。通常根据临床体征和下列检查结果可以确定诊断:①心电图;②超声心动图;③右心导管;④选择性左心室造影。
(5)法洛四联症(Tetralogy of Fallot):是一种常见的先天性心脏病,属发绀型畸形,包括肺动脉狭窄、室间隔缺损、主动脉骑跨和右心室肥大4种畸形。发绀是常见症状,通常患儿在1岁左右开始出现发绀,少数患儿肺动脉轻度狭窄,可不出现发绀;多数患儿在劳累后有一种特殊的体位——蹲踞位。部分患儿缺氧,出现发作性晕厥,表现为呼吸困难、活动耐力差。本病可根据体征及心电图、X线、超
声心动图等检查结果做出诊断。
(6)主动脉狭窄(Aortic stenosis):通常指一组阻碍血液由左心室流入升主动脉的先天性畸形。本病的临床表现与狭窄程度有关。大多数轻度狭窄者在儿童期没有症状,生长和发育均正常;仅少数婴儿或新生儿发生呼吸困难或心力衰竭。一般在10岁以后才逐渐出现明显的呼吸困难、晕厥及胸痛等,并随年龄的增长而加重,甚至可出现左心衰竭或猝死。狭窄严重者,儿童时期可出现症状,主要表现出冠状动脉供血不足以及大脑缺血的症状。该病主要通过体征和X线检查、心电图检查结果等确诊。
(7)完全性大动脉血管错位(Complete transposition of the great artery):指两条大动脉(主动脉和肺动脉)与相应的两个心室(左心室和右心室)连接异常的一类先天性心脏畸形。患儿出生时往往正常。约80%的病例出生后1~5天出现发绀,5~6周加重。有的甚至昏厥、呼吸困难、心力衰竭,大多数1周内死亡。大多数患者全身严重发绀,不喜蹲踞。实验室检查血氧饱和度在50%~60%之间。根据体征及心电图、心导管、心血管造影、超声心动图等检查结果可做出诊断。
(8)肺动脉狭窄(Pulmonary stenosis):是一种常见的先天性心脏病,右心室与肺动脉之间的通道狭窄,而室间隔完整。症状轻重与狭窄程度和右心代偿功能有关。轻度狭窄者可无症状;中度狭窄者,劳累可后感气急和疲劳;严重病例,随年龄增长逐渐出现右心衰而表现颈静脉怒张、肝大及腹水等征象;极严重狭窄者,约半数病例在新
生儿期即出现烦躁不安、心动过速等症状。可根据体征、心电图及X线检查结果等做出诊断。
(9)肺动静脉瘘(Pulmonary arterio venous fistula):是指肺内动脉和静脉间的异常直接沟通、血管曲张或形成海绵状血管瘤,肺动脉内的血液不经过肺的氧合而直接流入肺静脉。病情轻重取决于右向左分流量的多少。分流量小者,可无症状,当右向左分流量超过体循环量的30%时,将出现心悸、呼吸困难等症状。少数患者出现咯血、贫血以及继发性红细胞增多。可根据体征和X线检查结果等做出诊断。
(10)永存动脉干(Persistent truncus arteriosus):又名共同动脉干或共同主动脉-肺动脉干,是自心底部仅发出一根单独的大动脉,此大动脉干下仅有一组半月瓣。所有冠状动脉和周围动脉均由此唯一的大动脉干发出,在共同半月瓣下方都有室间隔缺损。临床表现主要取决于肺血流量的多少,几乎所有患者均有不同程度的发绀、呼吸困难、气急、乏力等和心力衰竭表现,常有反复呼吸道感染史。诊断主要依据超声心动图和心血管造影检查结果。
(11)心脏异位(Malposition of the heart):是指心脏在胸腔内的位臵异常,是由于在胚胎发育期心脏及其他内脏旋转异常而致的先天性畸形。一般根据连接心底至心尖的心脏轴线分3种类型:①右位心,为心脏轴线指向右;②中位心,为心脏轴线指向下;③左位心,为心脏轴线指向左。右位心又分镜面右位心和右旋心;左位心包括正常左位心及左旋心。心脏异位可以单独存在,因无血流动力学障碍,临床上多属偶然发现。在心脏异位的个案,判断心脏-心房位,可根据
心电图P肢、支气管的形态、肝及下腔静脉的位臵来定位;超声心动图、磁共振成像检查对心室和大动脉的关系能做出正确诊断。
(12)先天性二尖瓣闭锁(Congenital mitral atresia):是一种罕见的复杂心脏畸形。本病的主要病理改变是二尖瓣口部位呈肌性闭锁或膜性闭锁。几乎所有的患儿均有不同程度的发绀。发绀发生在出生时或出生后短期内,并呈进行性加重。无肺动脉狭窄个案,肺血多,发绀较轻。在左右心房间交通小或合并肺动脉狭窄个案,肺血少,发绀明显,常因肺静脉回流受阻而产生肺水肿、肺淤血。根据二尖瓣锁闭的临床表现,结合超声心电图、心导管及造影检查结果,不难做出诊断。
附件2.出生缺陷的描述要点
对难于准确做出诊断的出生缺陷,请详细描述其临床特征,提供各种医学检查结果。以下为部分畸形的描述要点,对多发畸形可针对每一具体畸形分别描述。
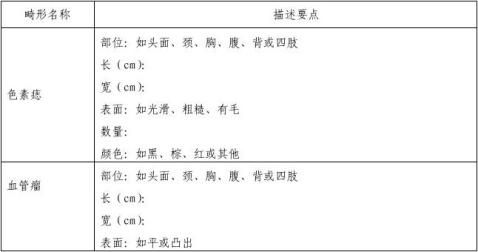
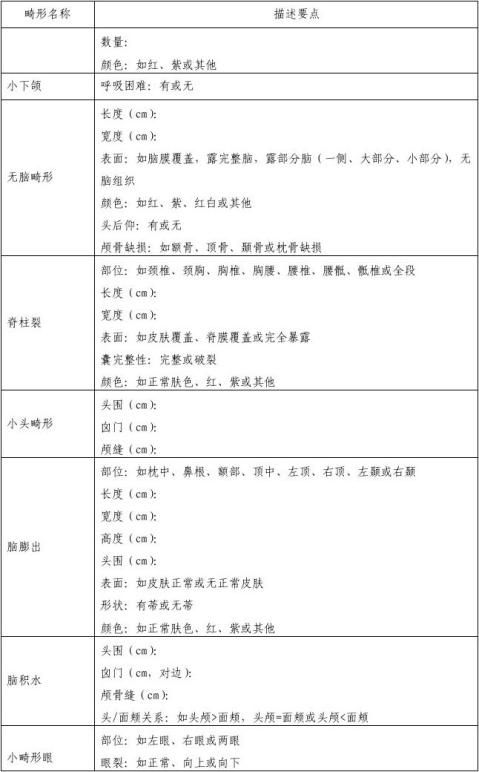
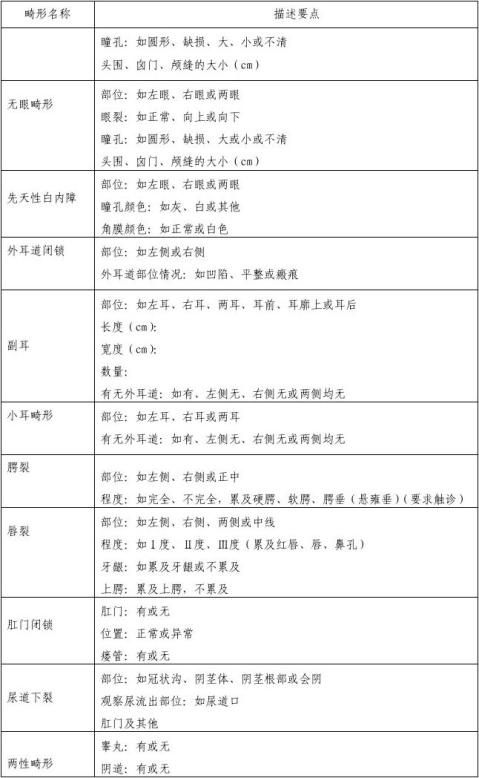
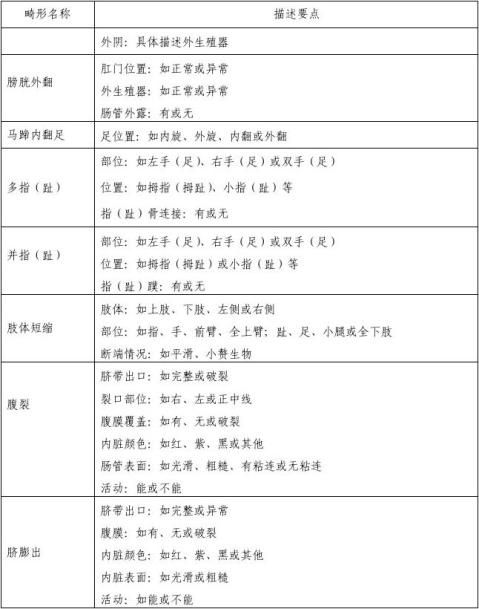
附件3.出生缺陷诊断流程

-
出生缺陷报告制度
出生缺陷报告制度一凡发现出生缺陷必须由妇保医生填写出生缺陷登记表二填报范围出生时有一种以上缺陷的围产儿均属填报范围三报告时间及程序…
-
新生儿出生缺陷报告制度及处理制度
温州新民妇产科医院新生儿出生缺陷报告制度及处理制度一建立新生儿出生缺陷登记簿如实填写围产儿出生缺陷登记表每月定期上报二分娩室发现出…
-
出生缺陷诊断制度(正式)
出生缺陷诊断制度出生缺陷的临床表现复杂多样涉及到各个系统各个器官部分缺陷出生时通过临床观察即可诊断但有些则必须借助丰富的相关专业知…
-
孕产妇死亡、5岁以下儿童死亡和出生缺陷报告制度
孕产妇死亡5岁以下儿童死亡和出生缺陷报告制度第一条为保障妇女儿童健康提高出生人口素质监测评估中国儿童发展纲要和中国妇女发展纲要主要…
-
出生缺陷报告制度
出生缺陷报告制度1妇幼保健机构及产科单位均应设专人负责出生缺陷信息收集上报工作2各助产单位对发生的出生缺陷儿认真记录填写出生缺陷儿…
-
出生缺陷报告和管理工作制度
出生缺陷报告和管理工作制度一意义为减少先天畸形儿的出生是提高我国人口素质的一个重要措施我国估计每年有3040万例体表先天畸形儿和8…
-
新生儿出生缺陷报告制度及处理制度
温州新民妇产科医院新生儿出生缺陷报告制度及处理制度一建立新生儿出生缺陷登记簿如实填写围产儿出生缺陷登记表每月定期上报二分娩室发现出…
-
出生缺陷报告制度
出生缺陷报告制度一凡发现出生缺陷必须由妇保医生填写出生缺陷登记表二填报范围出生时有一种以上缺陷的围产儿均属填报范围三报告时间及程序…
-
出生缺陷诊断制度(正式)
出生缺陷诊断制度出生缺陷的临床表现复杂多样涉及到各个系统各个器官部分缺陷出生时通过临床观察即可诊断但有些则必须借助丰富的相关专业知…
-
孕产妇死亡、5岁以下儿童死亡和出生缺陷报告制度
孕产妇死亡5岁以下儿童死亡和出生缺陷报告制度第一条为保障妇女儿童健康提高出生人口素质监测评估中国儿童发展纲要和中国妇女发展纲要主要…